Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Eun Ji Roh and Version 5 by Jason Zhu.
BIn research on various central nervous system injuries, bazedoxifene acetate, BZA, a third-generation selective estrogen receptor modulator (SERM), is an indole-based estrogen receptor ligand currently used to prevent and treat postmenopausal osteoporosis (BZA) has shown two main effects: neuroprotection by suppressing the inflammatory response and remyelination by enhancing oligodendrocyte precursor cell differentiation and oligodendrocyte proliferation.
- spinal cord injury
- secondary injury
- bazedoxifene
1. Introduction
RBZA, a third-generation selecently, Bazedoxifene acetate (tive estrogen receptor modulator (SERM), is an indole-based estrogen receptor ligand currently used to prevent and treat postmenopausal osteoporosis [17]. Recently, BZA) has been identified as a compelling remyelinating agent with a highly tolerable safety profile in a multiple sclerosis model [1][18]. Although SERMs have previously been implicated in remyelination and neuroprotection by targeting nuclear estrogen receptors (ERs), BZA has been found to promote remyelination independently of ERs [1][18]. Furthermore, research has revealed multiple neuroprotective effects of BZA in various central nervous system (CNS) injury models. Specifically, BZA has been shown to reduce ischemic brain damage and apoptosis by modulating the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK)1/2 and phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) signaling pathways [2][3][19,20]. Moreover, BZA has been found to attenuate impaired cognitive function and increased blood-brain barrier (BBB) permeability by modulating inflammation via MAPK pathway suppression in a traumatic brain injury (TBI) model [4][21]. In fact, pro-inflammatory microglia and the MAPK/ERK pathway are closely related to demyelination and remyelination processes in the injured CNS, undermining OPC survival and differentiation [5][6][7][22,23,24].
Similarly, evidence suggests that the main human estrogen, 17β-estradiol (E2) is a significant multi-active neuroprotective agent that may reduce inflammation and apoptosis, promote angiogenesis and myelination, and improve motor function after SCI [8][9][10][25,26,27].
2. Bazedoxifene Acetate (BZA) Promotes Survival of PC12 Neural Cells and Reduces In Vitro Nitric Oxide (NO) Production
To determine non-cytotoxic working concentrations of BZA, cell viability was measured by CCK-8 assay in PC12 cells. Compared to control and dimethyl sulfoxide (DMSO) group, lipopolysaccharide (LPS, 5 μg/mL) treatment significantly decreased cell viability from 100% to 44.03 ± 5.5%, but BZA 10 μM and BZA 20 μM treatments increased cell viability up to 74.25 ± 16.39% and 81.99 ± 23.1%, respectively (Figure 1A). However, the effect of BZA concentration was insignificant. The result indicates that both 10 μM and 20 μM BZA concentrations are optimal treatment concentrations for in vitro experiments.
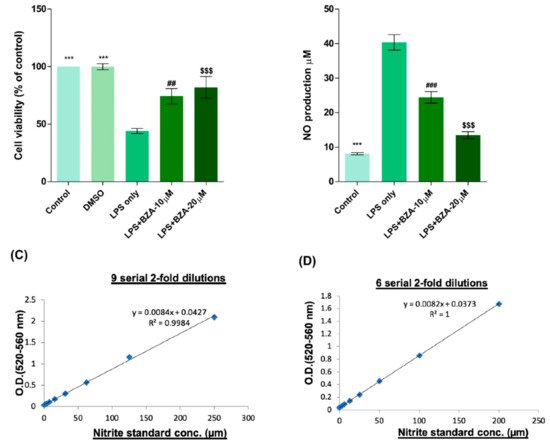
Figure 1. BZA promotes survival of PC12 neural cells and reduces in vitro NO production. Cell viability was significantly higher in LPS + BZA-treated cells compared with LPS-treated cells. NO production significantly decreased in response to 10 μM and 20 μM BZA treatment compared with LPS-treated cells. (A) Quantitative bar graph illustrating the percentage of live PC12 neural cells according to treatment group at 24 h following culture. (B) Quantitative bar graph illustrating inhibition of NO production in LPS-stimulated RAW264.7 cells treated with BZA. Standard curves to confirm the correlation between NO concentration and absorbance were obtained in two ways: (C) 9 serial 2-fold dilutions and (D) 6 serial 2-fold dilutions. Data are presented as mean ± SEM (n = 3 (B–D), n = 7 (A); performed in triplicate). *** p < 0.001 (LPS-treated vs. control, DMSO), ## p < 0.01, ### p < 0.001 (LPS-treated vs. LPS + BZA 10 μM), $$$ p < 0.001 (LPS-treated vs. LPS + BZA 20 μM), one-way ANOVA followed by the Bonferroni test.
The anti-inflammatory effect of BZA was evaluated in LPS-stimulated RAW 264.7 macrophage cells by a nitric oxide (NO) assay. The cells were treated with LPS and LPS + BZA (10 μM and 20 μM) and were examined 24 h after treatment. NO production increased steadily for 24 h after LPS treatment. The differences in NO production among the LPS-stimulated (non-treated), LPS + BZA 10 μM, and LPS + BZA 20 μM groups are presented in Figure 1B–D. As shown, NO production significantly decreased in the BZA-treated group in a dose-dependent manner compared with LPS-treated macrophage.
3. BZA Promotes In Vitro Inflammatory Resolution
The inflammatory response is a major pathologic feature of the secondary injury cascade after SCI. To evaluate the effect of BZA on the inflammatory response, the expression of pro-inflammatory (IL-6, TNF-α, CCL-2) and anti-inflammatory (IL-10) cytokines was measured in LPS-treated RAW 264.7 macrophages. As shown in Figure 2A–D, the expression levels of IL-6 (Figure 2A), TNF-α(Figure 2B), and CCL-2 (Figure 2C) were increased and that of IL-10 (Figure 2D) was decreased by LPS-induced inflammation. The increased levels of these pro-inflammatory cytokines driven by LPS were significantly impeded by BZA treatment, regardless of its concentration. However, the level of IL-10 significantly increased in the BZA-treated groups in a dose-dependent manner.
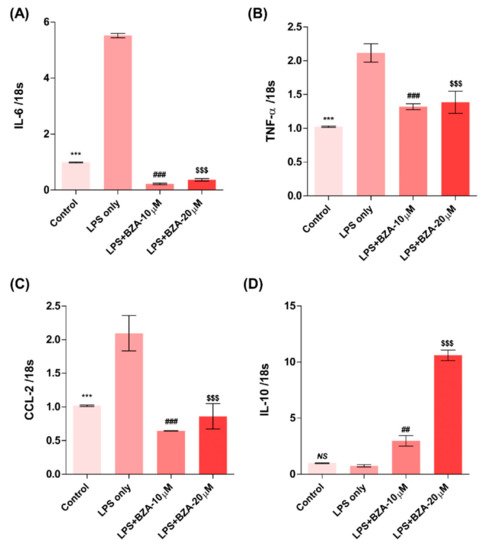
Figure 2. BZA promotes the resolution of inflammation in vitro. Total RNA was extracted from the LPS- and BZA (10 μM and 20 μM)-treated RAW 264.7 cells at 24 h following incubation (n = 3, performed in triplicate). The mRNA levels of proinflammatory cytokines including IL-6 (A), TNF-α (B), and CCL2 (C) and an anti-inflammatory cytokine, IL-10 (D), were measured. For time, 18 s was used as internal control for qRT-PCR. Data represent mean ± SEM (n = 3 (A,C), n = 4 (D), n = 6 (B); performed in triplicate). *** p< 0.001 (LPS-treated vs. control, DMSO), ## p < 0.01, ### p < 0.001 (LPS-treated vs. LPS + BZA 10 μM), $$$ p < 0.001 (LPS-treated vs. LPS + BZA 20 μM), NS = Not significant, one-way ANOVA followed by the Bonferroni test.
4. BZA Enhances Angiogenesis and Reduces In Vitro Blood-Spinal Cord Barrier (BSCB) Disruption
It is well established that blood–spinal cord barrier (BSCB) disruption after SCI allows blood cells to infiltrate the damaged parenchyma and exacerbates secondary injuries, such as focal edema, ischemia, focal hemorrhage, and inflammation [11][28]. Hence, maintaining BSCB permeability would induce a protective effect against secondary damage following SCI. The endothelial cells are connected by adhesion proteins and sealed by tight junction (TJ) proteins, which play an important role in maintaining the BSCB integrity [12][29]. In this study, we examined the changes of the TJ proteins including zonula occludens-1 (ZO-1) and occludin in a human cerebral microvascular endothelial cell line (hCMEC/D3) upon LPS-induced inflammation and evaluated the effects of BZA on these proteins with immunofluorescence imaging. As shown in Figure 3, LPS significantly decreased the levels of ZO-1 and occludin compared to the control. Subsequently BZA treatment significantly alleviated the reduction of ZO-1 and occludin expression. Similarly, disruption of the spinal cord vasculature is another factor that aggravates secondary injuries and reduces BSCB permeability after SCI [13][17]. Therefore, attempts to regulate angiogenic response and vascular stability have been made to promote neural regeneration and recovery [14][15][16][30,31,32]. To evaluate the effect of BZA on angiogenesis and vascular maintenance, the expression levels of angiopoietin-1 (ANGPT-1), von Willebrand factor (vWF), and α-smooth muscle actin (α-SMA) were determined in hCMEC/D3 cells. ANGPT-1 is crucial in limiting vascular permeability and controlling BSCB integrity, and eventually diminishing the inflammatory response by securing paracellular junctions [13][17]. Moreover, α-SMA is expressed in capillary pericytes, while vWF is a well-known angiogenic molecule [16][17][32,33]. Upon LPS treatment, the expression of ANGPT-1, vWF, and α-SMA decreased. In accordance with TJ proteins, these angiogenic proteins increased in response to BZA treatment (Figure 3). Taken together, the results demonstrate that BZA may contribute to the preservation of the BSCB integrity by inhibiting degradation of the TJ proteins and stabilizing vascularity.
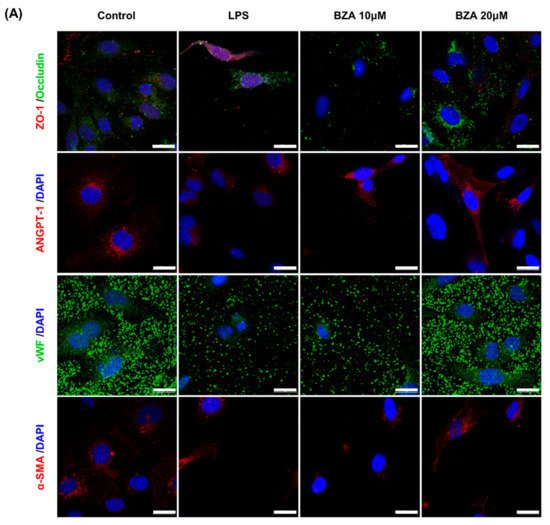
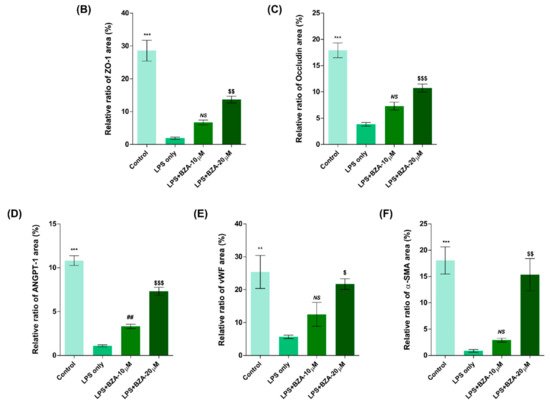
Figure 3. BZA increases angiogenesis and decreases BSCB destruction in vitro. (A) Representative immunofluorescent images (scale bar = 20 μm) of tight junction proteins such as ZO-1 and occludin and angiogenesis markers including ANGPT-1, vWF, and α-SMA examined in hCMEC/D3 cells. Quantitative fluorescence intensity for (B) ZO-1, (C) occludin, (D) ANGPT-1, (E) vWF, and (F) α-SMA. Data represent mean ± SEM (n = 3 (A,C), n = 4 (D), n = 6 (F); performed in triplicate). ** p < 0.01, *** p < 0.001 (LPS-treated vs. control, DMSO), ## p < 0.01 (LPS-treated vs. LPS + BZA 10 μM), $ p < 0.05, $$ p < 0.01, $$$ p < 0.001 (LPS-treated vs. LPS + BZA 20 μM), NS = Not significant, one-way ANOVA followed by the Bonferroni test.
5. BZA Attenuates Caspase-3-Induced Apoptotic Activity after SCI in Rats
Compared to the sham group, the injury-only group showed increased number of terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-positive cells in the lesion epicenter at 1 day post-injury (dpi). However, BZA treatment significantly reduced the TUNEL-positive cell counts compared to the injury-only group (Figure 4A,B). To directly examine the activity of the apoptosis executioner caspase-3, western blots were performed. The results showed higher expression of activated caspase-3 in the injury-only group (Figure 4C,D). Conversely, BZA attenuated SCI-induced caspase expression. Taken together, marked differences in TUNEL-positive cells and caspase-3 activity across the experimental groups demonstrate that BZA has neuroprotective role against apoptosis following acute SCI.
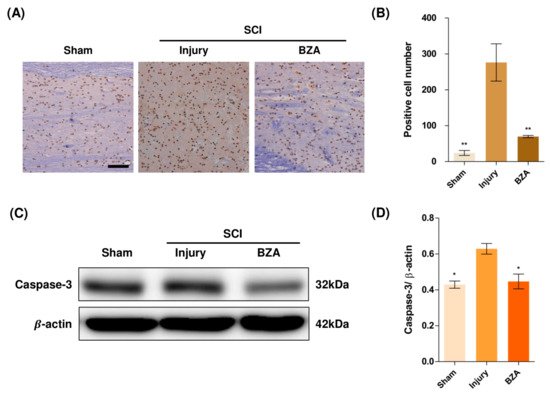
Figure 4. BZA attenuates caspase-3-induced apoptosis activity after SCI and suppresses the inflammatory response. (A) Representative images of the TUNEL assay observed in each group of spinal cord samples to examine apoptosis activity. (B) Graph result for positive cell number by quantitative analysis of TUNEL assay. To confirm the activity against apoptosis, western blot for caspase 3 was performed. (C) Representative images of western blot results for caspase 3 in the sham, injury, and BZA groups. (D) The graph of caspase 3 was measured to quantify the western blot results. All results of the graph confirmed the significance between the injury group and all other groups. * p < 0.05, ** p < 0.01, NS = Not significant. N = 3 (sham), 6 (injury), 5 (BZA) in the TUNEL assay and 3(C–D). TUNEL assay scale bar = 20 μm). Data represent mean ± SEM (in TUNEL, n = 3 (sham), 6 (injury), 5(BZA) and n = 3; (C,D) performed in triplicates). * p < 0.05, ** p < 0.01 (injury vs. sham, BZA), NS = Not significant, one-way ANOVA followed by the Bonferroni test.
6. BZA Downregulates the Phosphorylation of ERK and p38 Pathways Induced by SCI
Alterations in the activation of MAPKs were investigated in the rat spinal cord tissues at 48 h post-injury by Western blot analysis. The results indicated that BZA significantly attenuated the phosphorylation of ERK and p38 MAPKs (Figure 5). MAPKs are known to be upregulated in response to environmental stresses such as inflammatory stimuli and oxidative stress, playing a pivotal role in mediating SCI progression. Among MAPKs, the p38 pathway is considered to be crucial in the apoptosis network and in mobilizing major SCI-mediated proinflammatory cytokines such as IL-1β, TNF-α, and IL-6 [18][19][34,35].
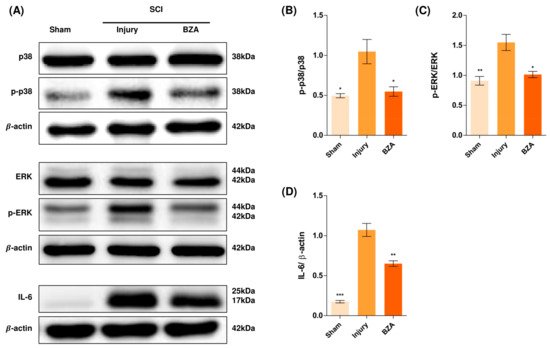
Figure 5. BZA downregulates the phosphorylation of ERK and p38 pathways. BZA decreases phosphorylation of ERK1/2 and p38, resulting in neuroprotection, OPC differentiation, and remyelination. (A) Representative images of western blots for p38, p-p38, ERK, p-ERK, IL-6. The graph was quantified as (B) p-p38/p38, (C) p-ERK/ERK and (D) IL-6. Western blot results of the graph confirmed the significance between the injury group and the sham and BZA groups. All data represent mean ± SEM (n = 3; performed in triplicate). * p < 0.05, ** p < 0.01, *** p < 0.001 (injury vs. sham, BZA), NS = Not significant, one-way ANOVA followed by the Bonferroni test.
Although mechanisms have not yet been fully elucidated, the ERK1/2 pathway is also known to participate in multiple secondary injury events, such as glutamate excitotoxicity, inflammation, apoptosis, and pain hypersensitivity [20][21][36,37]. Moreover, it has been reported that inhibition of ERK pathway promotes OL generation and recovery of demyelinating diseases and that acute strong induction of ERK in adulthood induces demyelination [6][22][23,38]. Furthermore, BZA has been shown to improve neurologic deficits in a TBI model and to reduce ischemic lesions in a stroke model by suppressing these MAPKs. Herein, we hypothesized that BZA would provoke similar outcomes in the SCI setting. Hence, as shown in Figure 5, phosphorylation of ERK and p38 significantly increased at 48 h after SCI, and this elevated expression was remarkably diminished upon BZA treatment.