Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Alexandre Harlé and Version 2 by Catherine Yang.
Damage-specific DNA-binding protein 2 (DDB2) was originally identified as a DNA damage recognition factor that facilitates global genomic nucleotide excision repair (GG-NER) in human cells. DDB2 also contributes to other essential biological processes such as chromatin remodeling, gene transcription, cell cycle regulation, and protein decay.
- DDB2
- DNA repair
- cancers
1. Introduction
DDB2 (damage-specific DNA-binding protein 2, also known as p48 subunit) is a 48-kDa protein exclusively localized in the nucleus of mammalian cells. DDB2 is ubiquitously present in human tissues, albeit differentially expressed. High DDB2 expression level has been described in liver, thymus, kidney, and testes, and a low expression level has been described in brain, lung, skin, heart, and muscles [1]. DDB2 is composed of seven WD40 repeat domains and a N-terminal helix–loop–helix motif [2]. The WD40 domains play a role as a module for sequence-specific protein–DNA or protein–protein interactions. Both domains appear critical for the biological functions of DDB2.
Numerous overlapping mechanisms have been found to be involved in the regulation of DDB2 expression. DDB2 basal expression is cell cycle-dependent in normal dividing cells with a DDB2 level that gradually increases in the mid-G1 phase and reaches a maximum at the G1/S boundary before dropping in the S-phase [3]. DDB2 transcription was shown to be transiently increased after UV-induced DNA damage in a p53-dependent manner [1]. The p53 protein cooperates with BRCA1 (Breast Cancer Associated protein 1) for its binding to the ddb2 promoter and inducing ddb2 transactivation [1] (Figure 1). The TAp63γ (Tumor protein 63 isoform gamma) isoform of the p63 protein that belongs to the p53 family and shares strong structural similarity with p53 is also able to activate DDB2 expression through recognition of the same region upstream of the transcription initiation site [4]. Structural analysis of the ddb2 promoter shows multiple Sp1 (Transcription factor Sp1) -specific binding sites as usually found in gene promoters with a G–C rich sequence lacking a TATA box and suggesting a critical role for Sp1 for the basal expression of DDB2 [5] (Figure 1). NF1 (Neurofibromin 1) and E2F (Transcription factor E2F) elements are also identified, although probably having a smaller impact on DDB2 regulation [5]. Moreover, DDB2 activity is finely adjusted through post-transcriptional and post-translational mechanisms. An IRES (internal ribosome entry site) element located at the 5′ end of DDB2 mRNA stimulates the translational process of DDB2 in stress conditions such as serum starvation or exposure to doxorubicin [6]. Moreover, the 3′ untranslated region (3′UTR) of DDB2 mRNA harbors an uracil-rich sequence enabling its prompt export to the cytoplasm and thus DDB2 translational upregulation. During the DNA repair process, the Cul4A (Cullin4A) protein regulates DDB2 protein lifespan by means of its ubiquitin ligase functions [3]. The PARylation (Protein poly ADP-ribosylation) of the DDB2 protein in a PARP1-dependent (Poly [ADP-ribose] polymerase 1) manner stabilizes DDB2 and delays its degradation [7]. A PIP (PCNA-interacting protein) box sequence located in the N-terminal region of DDB2 enables the interaction between DDB2 and PCNA (proliferating cell nuclear antigen) for DDB2 proteolytic degradation even in the absence of DNA damage [8][9][8,9].
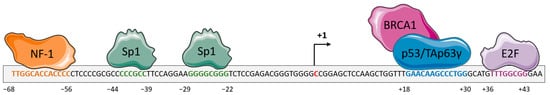
Figure 1. Schematic representation of the regulation of the gene encoding the DDB2 (Damage-specific DNA-binding protein 2) protein. The proximal promoter of ddb2 gene harbors response elements for the transcription factors NF-1 (Neurofibromin 1) (orange) and Sp1 (Transcription factor Sp1) (green) upstream of the transcription initiation site. The proximal promoter also contains a response element for p53, in association with BRCA1 (Breast cancer type 1 susceptibility protein), or Tap63γ (Tumor protein 63 isoform gamma) (blue) proteins and the E2F (Transcription factor E2F) transcription factor downstream of the transcription initiation site. The binding of these proteins leads to the regulation of ddb2 gene expression.
2. DDB2: A New Potent Tumor Suppressor?
DDB2 was first considered as a novel tumor suppressor based on the findings that mutations in the ddb2 gene result in an impairment in DDB2–DNA or DDB2–DDB1 interactions and subsequent NER activity defects [2]. Such deficiencies are observed in a subset of Xeroderma pigmentosum hereditary disease (Xeroderoma pigmentosum group E, XP-E) that displays an extreme sensitivity to UV radiation and a high predisposition to skin cancers [7]. Moreover, DDB2-deficient mice are prone to developing a wide panel of tumors, even in the absence of UV exposition [10][19]. Several studies highlighted an altered DDB2 expression, compared to non-malignant tissues, in many types of cancers [11][20], including prostate [12][21], colorectal [13][14][22,23], skin [15][24], head and neck [16][25], and ovarian [17][26] cancers. Furthermore, a correlation between low DDB2 expression level and poor outcomes was established among patients with melanoma, colon, ovarian, lung, breast, brain or head, and neck cancers, suggesting a critical role for DDB2 in tumor suppression [13][16][17][18][22,25,26,27].
3. DDB2 Has a Dual Activity on Cancer Cell Proliferation
High levels of DDB2 protein and mRNA are reported in ER (Estrogen receptor)-positive and non-invasive breast cancer models compared to ER-negative aggressive breast cancer cells and mammary non-malignant cells [19][16]. Enhanced DDB2 expression in DDB2-low level models upregulates in vitro cancer cell growth rate and clonogenicity. Such effects are abrogated by DDB2 knockdown in DDB2-overexpressed breast cancer models, suggesting the oncogenic role of DDB2 in mammary cancer cell growth [19][16]. DDB2 facilitates cell cycle progression, especially the entry in the S-phase, through the binding of the C-terminal domain and the co-activation of the transcription factor E2F1 [3]. By means of E2F1 transcriptional abilities, DDB2 indirectly regulates the expression of key genes involved in DNA replication and G1/S transition. The stimulating effect of DDB2 on cancer cell growth also involves the downregulation of manganese superoxide dismutase (MnSOD) [19][16]. MnSOD is a mitochondrial enzyme that detoxifies reactive oxygen species (ROS) to protect cells from oxidative damage. DDB2 interacts with the proximal SOD2 (Superoxide dismutase 2) promoter, resulting in the loss of H3 histone acetylation, and in the recruitment of the AP-2 transcription factor, which is well known in the repression of the SOD2 gene and downregulation of the encoded MnSOD [20][28]. By this mechanism, DDB2 attenuates the elimination of ROS that are known to activate several signaling pathways involved in breast cancer cell growth [20][28].
In contrast, DDB2 has shown antiproliferative properties in ovarian and prostate cancers in vitro. In an ovarian cancer cell model, DDB2 is reported to negatively regulate NEDD4L (Neural precursor cell expressed developmentally downregulated gene 4-like) cellular levels by inducing histone H3 trimethylation at the NEDD4L promoter region [21][29]. This limits the NEDD4L-dependent proteolytic degradation of the effector proteins SMAD2 (Mothers against decapentaplegic homolog) and SMAD3 and enhances the TGF-β (Transforming growth factor beta) signal transduction downstream, finally contributing to the inhibition of cancer cell proliferation [21][29].
NRIP/DCAF6 (Nuclear receptor-interacting protein 1/DDB1- and CUL4-associated factor 6) and DDB2 proteins are both members of the DDB1 and CUL4-associated factors DCAF family and androgen receptor (AR)-interacting proteins that physiologically compete to maintain AR expression level. DDB2 mediates the contact between AR and CUL4A–DDB1 E3 ligase complex for AR ubiquitination and proteasomal degradation in a p53-independent manner [22][30], while NRIP displaces DDB2 from the complex and stabilizes AR [12][21]. As the DDB2 expression level is found to be lower in prostate cancer tissues compared to non-neoplastic ones, this could interfere with AR homeostasis and induce subsequent AR-dependent prostate cancer growth.
Deregulation in the Wnt signaling pathway is usually co-opted during colon cancer development and is sought to be a driver event in this process. Negative regulatory effects of DDB2 in the Wnt signaling are mediated by the recruitment of β-catenin and the H3K27 methylase EZH2 (Enhancer of zeste homolog 2) to Rnf43 (Ring Finger protein 43) promoter. This facilitates the activation of the RNF43 enzyme that eliminates Wnt receptors at the cell surface and downregulates Wnt signaling in colorectal cancer cells [23][31].
4. DDB2 Affects Cancer Stem Cell Populations
Some studies reveal the protective role of DDB2 for ovarian cancer progression and recurrence by limiting the cancer stem cell (CSC) population. High levels of DDB2 are shown to halt the formation of ovarian tumor xenografts in vivo [17][26]. DDB2 exerts this regulatory function through the increase of IκBα protein levels, which results in the repression of the NF-κB signaling pathway and impinges the capacity of CSC to self-renew [17][26]. DDB2 also represses ovarian cancer cell dedifferentiation by inhibiting the transcription of aldehyde dehydrogenase 1 family member A1 (ALDH1A1) [24][39]. The impact of DDB2 on cancer stem cell populations is still under investigation in other cancer models and need to be explored.