This reviews describes the requirement of ion channel function for viral infection. Also discussed is how viral infections can result in acquired channelopathies by altering ion channels. Ion channel modulators may be useful in treating both of these aspects.
- Ion channel, virus, channelopathy
- virus-host interactions
1. Introduction
The human “channelome” contains over 300 known channels [1] that selectively and rapidly transport ions across biological membranes in response to specific stimuli. Ion channels are present on the plasma membranes and organelles of all cells, where they regulate organelle ion homeostasis, mitochondrial function, inflammasome activation, action potential firing, membrane potential, cell volume, and autophagy [2][3][4][5]. Given their importance, it follows that their dysfunctions leads to human diseases, termed channelopathies [6]. These include disease states of the nervous [2], musculoskeletal [5], cardiovascular [7], and immune systems [8]. This has motivated research on compounds that can modulate ion channel activity; ~19% of current FDA-approved drugs are ion channel modulators, second only to drugs targeting G-protein coupled receptors [9][10].
The human “channelome” contains over 300 known channels [1] that selectively and rapidly transport ions across biological membranes in response to specific stimuli. Ion channels are present on the plasma membranes and organelles of all cells, where they regulate organelle ion homeostasis, mitochondrial function, inflammasome activation, action potential firing, membrane potential, cell volume, and autophagy [2,3,4,5]. Given their importance, it follows that their dysfunctions leads to human diseases, termed channelopathies [6]. These include disease states of the nervous [2], musculoskeletal [5], cardiovascular [7], and immune systems [8]. This has motivated research on compounds that can modulate ion channel activity; ~19% of current FDA-approved drugs are ion channel modulators, second only to drugs targeting G-protein coupled receptors [9,10].Many viruses encode their own ion channels [11][12][13] termed “viroporins,” highlighting the importance of ionic balance during viral infection. This field has spurred intense research and several drugs that target viroporins have emerged (reviewed in [11]). More recent evidence highlights how viruses can regulate and/or depend on the ion channels expressed by host cells, highlighting them as new host targets for therapeutic intervention (reviewed by Hover et al., 2017) [14].
2. Ion Channels Involved in Viral Entry
2.1. Ca
2+
Channels and Viral Entry
The involvement of Ca
2+ channels during viral entry is now well-documented [15]. Fujioka et al. showed that influenza virus (IAV) hemagglutinin (HA) triggers intracellular [Ca
2+] oscillations that are required for viral infection [16]. The initial modulation of Ca
2+
2+
2+
2+
V
V
V
2+ blocker, highlighting the potential of these compounds for drug repurposing.
Ebola virus (EBOV) also requires Ca
2+
2+ channel 2 (TPC2) [17]. To further characterise this pathway, Penny et al. expanded the pharmacology of TPCs using a virtual screen of ~1500 FDA-approved drugs. All identified TPC modulators were cross-referenced with two recent anti-EBOV screens, with four dopamine receptor antagonists and five oestrogen receptor modulators identified. As such, it was reasoned that these drugs exert their inhibitory effects on EBOV through the blockade of TPCs (Figure 1E), subsequently confirmed through EBOV virus-like particle (VLP) assays [18]. Das et al. further characterised the role of Ca
2+
2+ and pH synergistically induce a conformational change in the EBOV glycoprotein GP2 (a key mediator of receptor binding and viral entry) to form a reversible intermediate state primed for NPC1 binding. NPC1 binding then further promotes the conformational transition into a fusion-ready “primed” state of invading EBOV virions [19].
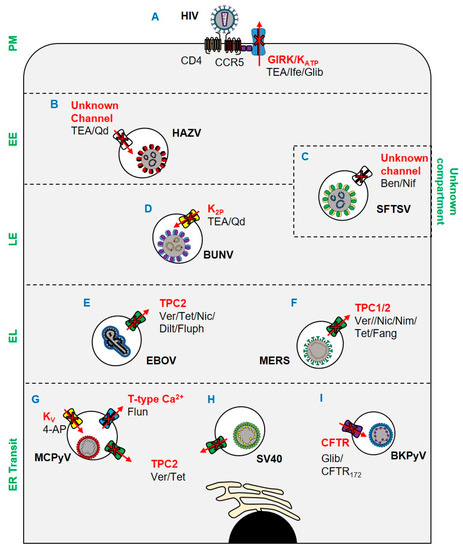
Figure 1.
A
ATP
B
C
D
2P
E
F
G
H
V
I) ER trafficking of BKPyV is CFTR dependent and susceptible to blockade by CFTR172 and glibenclamide. Key: PM: plasma membrane; EE: early endosome; LE: late endosome; EL: endolysosome. Ver: verapamil; Tet: tetrandrine; Nic: nicardipine; Dilt: diltiazem; Fluph: fluphenazine; Fang: fangchinoline; Nim: nimodipine; Nif: nifedipine; TEA: tetraethylammonium; Qd: quinidine; Ife: ifenprodil; Glib: glibenclamide; 4-AP: 4-aminopyridine.
Of importance to the current pandemic, it has been shown that Middle East respiratory syndrome coronavirus (MERS) [20] is dependent on TPCs to escape endosomes [21]. As an enveloped virus, MERS must fuse its envelope with host membranes to enter cells. Following receptor attachment, MERS particles can fuse at either the cell surface or intracellularly in the endosomal network. Fusion is mediated by the proteolytic cleavage of the viral spike (S) protein at its S1/S2 site. At the surface of the cell, this proteolytic event is facilitated by TMPRSS2, which in turn precludes exposure of the fusion loop and coalescence of host and viral membranes [22]. Alternatively, fusion can occur in late endosomes following translocation through the endocytic network and proteolytic processing by proprotein convertases, including furin, in a process regulated by Ca
2+ [23][24]. In studies by Gunaratne et al., genetic silencing of TPC1 and TPC2 prevented the entry of pseudotyped MERS (Figure 1F). The dependence of MERS upon TPCs was further demonstrated through its inhibition by tetrandrine and fangchinoline (TPC inhibitors), which prevented a post-internalisation but pre-fusion entry event. The mechanism through which TPC blockade inhibited MERS was multi-faceted: TPC1 and TPC2 silencing impaired furin activity, whilst pharmacological and genetic inhibition of TPC1 impaired endosomal motility. Of note, the related SARS-CoV-2, the causative agent of COVID-19 [25][26], was similarly inhibited by TPC blockade. Specifically, treatment of cells with tetrandrine reduced the entry of a lentiviral vector pseudotyped with the SARS-CoV-2 S (spike) [27].
The bunyavirus severe fever with thrombocytopenia syndrome virus (SFTSV) is an emerging arbovirus with fatality rates of 12–50% and the potential to cause future pandemics [28]. Using a library of FDA-approved drugs, the Ca
2+ channel blockers benidipine hydrochloride and nifedipine were shown to inhibit SFTSV infection (Figure 1C), with in vivo activity confirmed using C57BL/6 and humanised mouse models. Through a retrospective analysis of human SFTSV cases, clinical evidence of the efficacy of nifedipine as an anti-SFTSV therapeutic was also demonstrated. A cohort of patients receiving nifedipine prior to and during hospital admission showed enhanced viral clearance and reduced frequency of neurological syndromes, often associated with fatal outcomes [29][30]. The fatality rate of patients receiving nifedipine was reduced 5-fold compared to untreated patients, which corresponded to abnormal serum Ca
2+
2+ channels were subsequently shown to be during virus internalisation and genome replication.
2.2. K
+
Channels and Viral Entry
The involvement of K
+
+
+
2P) inhibited the early stages of the BUNV lifecycle (Figure 1D) [31]. Subsequent work identified both acidic pH and K
+ in endosomes as crucial biochemical cues for the endosomal escape of BUNV [32]. Similar studies in HAZV highlighted a dependence on K
+
+ primarily accumulated in cholesterol-rich endosomes (Figure 1B) [32][33][34]. The K
+
+
+
+
+
+
+ therefore drives viral uncoating and expedites IAV infection [35].
Figure 2.
A
i
A
ii
+
A
iii
B
i
B
ii
B
iii
+
+
B
iv)) At low pH, a conformational change in HA promotes fusion and RNP release.
Recent work also highlights a requirement for K
+ channels during human immunodeficiency virus (HIV) infection. Using pharmacological approaches (Figure 1A) [36] HIV entry could be blocked with ifenprodil and the broad spectrum K
+ channel blocker tetraethylammonium (TEA). Khan et al. also showed that the pharmacological activation of the endolysosome-resident transient receptor potential mucolipin 1 channel (TRPML1) enhanced the degradation of HIV-Tat (a multi-function viral protein involved in transcription, splicing, capping, and translation), which in turn reduced the transition from viral latency [37]. TRPML1 activates large conductance Ca
2+-activated potassium (BK) channels in endosomes [38], implying that this channel is required for HIV infection.
The reliance of viruses upon ion channels is not restricted to RNA viruses. It was recently shown that both K
+
2+ channels are important host factors for polyomavirus infection [39]. Using a panel of ion channel modulators, the entry of Merkel cell polyomavirus (MCPyV), the causative agent of Merkel cell carcinoma (MCC), was shown to be sensitive to 4-aminopyridine (4-AP), a blocker of voltage-gated K
+
V
2+
2+ channels involved in polyomavirus entry were further explored, revealing a requirement for transient (T-type, low-voltage activated) channel family members in MCPyV infection but not SV40. Tetrandrine, a TPC blocker, restricted both viruses. The role of TPCs was found to be during endoplasmic reticulum (ER) disassembly and/or ER docking for SV40 [40], which may be explained by the recent demonstration that Ca
2+ ions mediate the stabilization of SV40 capsids and contribute to its disassembly [41].
2.3. Cl
−
Channels and Viral Entry
BK polyomavirus (BKPyV) is a potentially fatal pathogen in patients undergoing solid organ transplantation. Panou et al. demonstrated that the pharmacological and genetic disruption of the cystic fibrosis transmembrane conductance regulator (CFTR) Cl
−
172 and glibenclamide, combined with the assessment of exposure of VP2/VP3 minor capsid proteins, indicated a role for CFTR in the trafficking of BKPyV to the ER. Whilst the mechanism of CFTR involvement in BKPyV ER trafficking remains unclear, it is hypothesised that the channel may be important in the acidification and ER docking of BKPyV-containing endosomes. CFTR has been implicated in the fusion of endosomes [43], and co-localises with vacuolar ATPase (vATPase) to provide the counter-charge during endosomal acidification [44].
References
- Luzio, J.P.; Bright, N.; Pryor, P. The role of calcium and other ions in sorting and delivery in the late endocytic pathway. Biochem. Soc. Trans. 2007, 35, 1088–1091. [Google Scholar] [CrossRef]
- Gerasimenko, O.V.; Tepikin, A.V.; Petersen, O.H.; Gerasimenko, O.V. Calcium uptake via endocytosis with rapid release from acidifying endosomes. Curr. Biol. 1998, 8, 1335–1338. [Google Scholar] [CrossRef]
- Hover, S.; Foster, B.; Fontana, J.; Kohl, A.; Goldstein, S.A.; Barr, J.N.; Mankouri, J. Bunyavirus requirement for endosomal K+ reveals new roles of cellular ion channels during infection. PLoS Pathog. 2018, 14, e1006845. [Google Scholar] [CrossRef]
- Staring, J.; Raaben, M.; Brummelkamp, T.R. Viral escape from endosomes and host detection at a glance. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef]
- Fujioka, Y.; Nishide, S.; Ose, T.; Suzuki, T.; Kato, I.; Fukuhara, H.; Fujioka, M.; Horiuchi, K.; Satoh, A.O.; Nepal, P.; et al. A sialylated voltage-dependent Ca2+ channel binds hemagglutinin and mediates influenza a virus entry into mammalian cells. Cell Host Microbe 2018, 23, 809–818. [Google Scholar] [CrossRef]
- Stauffer, S.; Feng, Y.; Nebioglu, F.; Heilig, R.; Picotti, P.; Helenius, A. Stepwise priming by acidic pH and a high K+ concentration is required for efficient uncoating of influenza a virus cores after penetration. J. Virol. 2014, 88, 13029–13046. [Google Scholar] [CrossRef] [PubMed]
- Simmons, J.A.; D’Souza, R.S.; Ruas, M.; Galione, A.; Casanova, J.E.; White, J.M. Ebolavirus glycoprotein directs fusion through NPC1+ endolysosomes. J. Virol. 2015, 90, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Penny, C.J.; Vassileva, K.; Jha, A.; Yuan, Y.; Chee, X.; Yates, E.; Mazzon, M.; Kilpatrick, B.S.; Muallem, S.; Marsh, M.; et al. Mining of Ebola virus entry inhibitors identifies approved drugs as two-pore channel pore blockers. Biochim. Biophys. Acta Mol. Cell Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Gunaratne, G.S.; Yang, Y.; Li, F.; Walseth, T.F.; Marchant, J.S. NAADP-dependent Ca2+ signaling regulates Middle East respiratory syndrome-Coronavirus pseudovirus translocation through the endolysosomal system. Cell Calcium 2018, 75, 30–41. [Google Scholar] [CrossRef]
- Li, H.; Zhang, L.K.; Li, S.F.; Zhang, S.F.; Wan, W.W.; Zhang, Y.L.; Xin, Q.L.; Dai, K.; Hu, Y.Y.; Wang, Z.B.; et al. Calcium channel blockers reduce severe fever with thrombocytopenia syndrome virus (SFTSV) related fatality. Cell Res. 2019, 29, 739–753. [Google Scholar] [CrossRef]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Hover, S.; King, B.; Hall, B.; Loundras, E.A.; Taqi, H.; Daly, J.; Dallas, M.; Peers, C.; Schnettler, E.; McKimmie, C.; et al. Modulation of potassium channels inhibits bunyavirus infection. J. Biol. Chem. 2016, 291, 3411–3422. [Google Scholar] [CrossRef]
- Punch, E.K.; Hover, S.; Blest, H.T.; Fuller, J.; Hewson, R.; Fontana, J.; Mankouri, J.; Barr, J.N. Potassium is a trigger for conformational change in the fusion spike of an enveloped RNA virus. J. Biol. Chem. 2018, 293, 9937–9944. [Google Scholar] [CrossRef]
- Charlton, F.W.; Hover, S.; Fuller, J.; Hewson, R.; Fontana, J.; Barr, J.N.; Mankouri, J. Cellular cholesterol abundance regulates potassium accumulation within endosomes and is an important determinant in bunyavirus entry. J. Biol. Chem. 2019, 294, 7335–7347. [Google Scholar] [CrossRef]
- Dubey, R.C.; Mishra, N.; Gaur, R. G protein-coupled and ATP-sensitive inwardly rectifying potassium ion channels are essential for HIV entry. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef]
- Dobson, S.J.; Mankouri, J.; Whitehouse, A. Identification of potassium and calcium channel inhibitors as modulators of polyomavirus endosomal trafficking. Antiviral Res. 2020, 104819. [Google Scholar] [CrossRef] [PubMed]
- Panou, M.M.; Antoni, M.; Morgan, E.L.; Loundras, E.A.; Wasson, C.W.; Welberry-Smith, M.; Mankouri, J.; Macdonald, A. Glibenclamide inhibits BK polyomavirus infection in kidney cells through CFTR blockade. Antiviral Res. 2020, 178, 104778. [Google Scholar] [CrossRef] [PubMed]
- Das, D.K.; Bulow, U.; Diehl, W.E.; Durham, N.D.; Senjobe, F.; Chandran, K.; Luban, J.; Munro, J.B. Conformational changes in the Ebola virus membrane fusion machine induced by pH, Ca2+, and receptor binding. PLoS Biol. 2020, 18, e3000626. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Hui, D.S.; Perlman, S. Middle east respiratory syndrome. Lancet 2015, 386, 995–1007. [Google Scholar] [CrossRef]
- Park, J.E.; Li, K.; Barlan, A.; Fehr, A.R.; Perlman, S.; McCray, P.B.; Gallagher, T. Proteolytic processing of middle east respiratory syndrome coronavirus spikes expands virus tropism. Proc. Natl. Acad. Sci. USA 2016, 113, 12262–12267. [Google Scholar] [CrossRef]
- Mille, J.K.; Whittaker, G.R. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc. Natl. Acad. Sci. USA 2014, 111, 15214–15219. [Google Scholar] [CrossRef]
- Ruas, M.; Rietdorf, K.; Arredouani, A.; Davis, L.C.; Lloyd-Evans, E.; Koegel, H.; Funnell, T.M.; Morgan, A.J.; Ward, J.A.; Watanabe, K.; et al. Purified TPC isoforms form NAADP receptors with distinct roles for Ca2+ signaling and endolysosomal trafficking. Curr. Biol. 2010, 20, 703–709. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Li, H.; Lu, Q.B.; Xing, B.; Zhang, S.F.; Liu, K.; Du, J.; Li, X.K.; Cui, N.; Yang, Z.D.; Wang, L.Y.; et al. Epidemiological and clinical features of laboratory-diagnosed severe fever with thrombocytopenia syndrome in China, 2011–17: A prospective observational study. Lancet Infect. Dis. 2018, 18, 1127–1137. [Google Scholar] [CrossRef]
- Bao, C.J.; Guo, X.L.; Qi, X.; Hu, J.L.; Zhou, M.H.; Varma, J.K.; Cui, L.B.; Yang, H.T.; Jiao, Y.J.; Klena, J.D.; et al. A family cluster of infections by a newly recognized bunyavirus in Eastern China, 2007: Further evidence of person-to-person transmission. Clin. Infect. Dis. 2011, 53, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Lakpa, K.L.; Halcrow, P.W.; Afghah, Z.; Miller, N.M.; Geiger, J.D.; Chen, X. BK channels regulate extracellular Tat-mediated HIV-1 LTR transactivation. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Zhong, X.Z.; Zou, Y.; Zhang, Z.; Toro, L.; Dong, X.P. BK channels alleviate lysosomal storage diseases by providing positive feedback regulation of lysosomal Ca2+ release. Dev. Cell 2015, 33, 427–441. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, Y.; Kolokoltsov, A.A.; Chen, C.C.; Tidwell, M.W.; Bauta, W.E.; Klugbauer, N.; Grimm, C.; Wahl-Schott, C.; Biel, M.; Davey, R.A. Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science 2015, 347, 995–998. [Google Scholar] [CrossRef] [PubMed]
- Kawano, M.A.; Xing, L.; Tsukamoto, H.; Inoue, T.; Handa, H.; Cheng, R.H. Calcium bridge triggers capsid disassembly in the cell entry process of simian virus 40. J. Biol. Chem. 2009, 284, 34703–34712. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, N.A. Intracellular CFTR: Localization and function. Physiol. Rev. 1999, 79, S175–S191. [Google Scholar] [CrossRef]
- Collaco, A.M.; Geibel, P.; Lee, B.S.; Geibel, J.P.; Ameen, N.A. Functional vacuolar ATPase (V-ATPase) proton pumps traffic to the enterocyte brush border membrane and require CFTR. Am. J. Physiol. Cell Physiol. 2013, 305, C981–C996. [Google Scholar] [CrossRef]