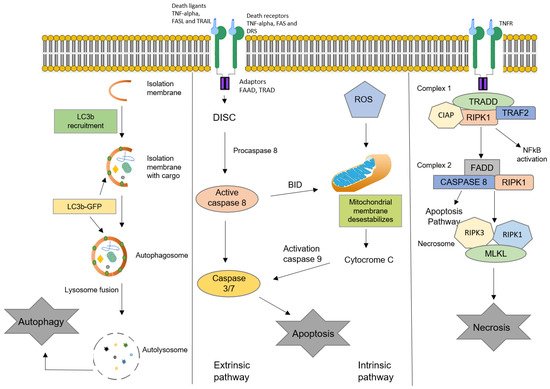
Oxysterols are oxidized derivatives of cholesterol produced by enzymatic activity or non-enzymatic pathways (auto-oxidation). The oxidation processes lead to the synthesis of about 60 different oxysterols. Several oxysterols have physiological, pathophysiological, and pharmacological activities. The effects of oxysterols on cell death processes, especially apoptosis, autophagy, necrosis, and oxiapoptophagy (a complex mode of cell death characterized by ROS overproduction (“oxi-”), apoptosis induction, (“-apopto”), and autophagy (“-phagy”)), as well as their action on cell proliferation, are reviewed here. These effects, also observed in several cancer cell lines, could potentially be useful in cancer treatment. The effects of oxysterols on cell differentiation are also described. Among them, the properties of stimulating the osteogenic differentiation of mesenchymal stem cells while inhibiting adipogenic differentiation may be useful in regenerative medicine.
- apoptosis
- autophagy
- cell death
- differentiation
- oxysterol
- Oxiapoptophagy
- oxiapoptophagy
1. Introduction
2. Oxysterols
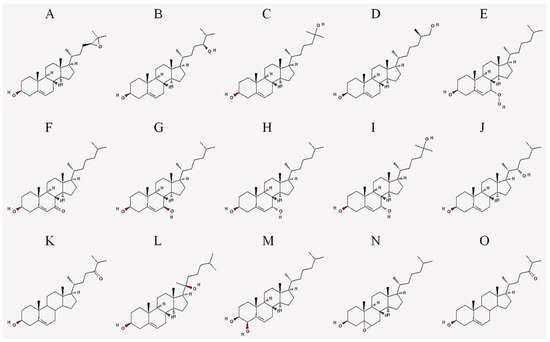
| Abbreviation | Common Name | IUPAC Name |
|---|---|---|
| 24,25-EC | 24(S),25-epoxycholesterol | (3S,8S,9S,10R,13R,14S,17R)-17-[(2R)-4-[(2S)-3,3-dimethyloxiran-2-yl]butan-2-yl]-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-ol |
| 24-HC | 24(S)-hydroxycholesterol | (3S,8S,9S,10R,13R,14S,17R)-17-[(2R,5S)-5-hydroxy-6-methylheptan-2-yl]-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-ol |
| 25-HC | 25-hydroxycholesterol | (3S,8S,9S,10R,13R,14S,17R)-17-[(2R)-6-hydroxy-6-methylheptan-2-yl]-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-ol |
| 27-HC | 27-hydroxycholesterol | (3S,8S,9S,10R,13R,14S,17R)-17-[(2R,6R)-7-hydroxy-6-methylheptan-2-yl]-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-ol |
| 7-OOHC | 7-hydroperoxycholesterol | (3S,8S,9S,10R,13R,14S,17R)-7-hydroperoxy-10,13-dimethyl-17-[(2R)-6-methylheptan-2-yl]-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-ol |
| 7α-HC | 7α-hydroxycholesterol | (3S,7S,8S,9S,10R,13R,14S,17R)-10,13-dimethyl-17-[(2R)-6-methylheptan-2-yl]-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthrene-3,7-diol |
| 7β-HC | 7β-hydroxycholesterol | (3S,4R,8S,9S,10R,13R,14S,17R)-10,13-dimethyl-17-[(2R)-6-methylheptan-2-yl]-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthrene-3,4-diol |
| 7-KC | 7-ketocholesterol | (3S,8S,9S,10R,13R,14S,17R)-3-hydroxy-10,13-dimethyl-17-[(2R)-6-methylheptan-2-yl]-1,2,3,4,8,9,11,12,14,15,16,17-dodecahydrocyclopenta[a]phenanthren-7-one |
| 7α,25-DHC | 7α,25-dihydroxycholesterol | (3S,7S,8S,9S,10R,13R,14S,17R)-17-[(2R)-6-hydroxy-6-methylheptan-2-yl]-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthrene-3,7-diol |
| 7β,27-DHC | 7β,27-dihydroxycholesterol | (3S,7R,8S,9S,10R,13R,14S,17R)-17-[(2R)-7-hydroxy-6-methylheptan-2-yl]-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthrene-3,7-diol |
| 22-HC | 22(S)-hydroxycholesterol | (3S,8S,9S,10R,13S,14S,17R)-17-[(2S,3S)-3-hydroxy-6-methylheptan-2-yl]-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-ol |
| 20-HC | 20(S)-hydroxycholesterol | (3S,8S,9S,10R,13S,14S,17S)-17-[(2S)-2-hydroxy-6-methylheptan-2-yl]-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-3-ol |
| 4β-HC | 4β-hydroxycholesterol | (3S,4R,8S,9S,10R,13R,14S,17R)-10,13-dimethyl-17-[(2R)-6-methylheptan-2-yl]-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthrene-3,4-diol |
| 7,25-DHC | 7,25-dihydroxycholesterol | (3S,8S,9S,10R,13R,14S,17R)-17-[(2R)-6-hydroxy-6-methylheptan-2-yl]-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthrene-3,7-diol |
| 5,6-EC | 5,6-epoxycholesterol | (1S,2R,5S,11S,12S,15R,16R)-2,16-dimethyl-15-[(2R)-6-methylheptan-2-yl]-8-oxapentacyclo[9.7.0.02,7.07,9.012,16]octadecan-5-ol |
| 24-OXO | 24-oxocholesterol | (6R)-6-[(3S,10R,13R,17R)-3-hydroxy-10,13-dimethyl-2,3,4,7,8,9,11,12,14,15,16,17-dodecahydro-1H-cyclopenta[a]phenanthren-17-yl]-2-methylheptan-3-one |
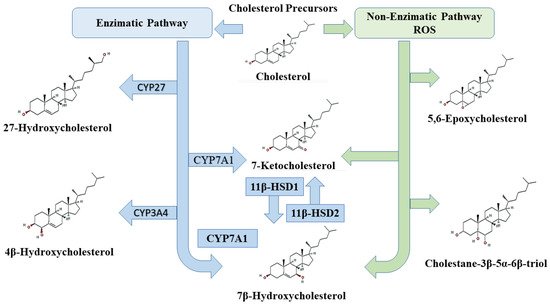
3. Oxysterols and Cell Death