Human immunodeficiency virus type-1 (HIV-1) can undergo either a lytic pathway to cause productive systemic infection or enter a latent state in which the integrated provirus remains transcriptionally silent for decades.
The ability to latently infected T-cells enables HIV-1 to establish persistent infections in resting memory CD4+ T-lymphocytes which become reactivated following disruption or cessation of intensive drug therapy.
Maintenance of viral latency occurs through epigenetic and non-epigenetic mechanisms.
Epigenetic mechanisms of HIV latency regulation involve deacetylation and methylation of histone proteins within Nucleosome 1 (nuc -1) at the viral long terminal repeats (LTR) such that inhibition of histone deacetyltransferase and histone lysine methyltransferase activities, respectively, reactivates HIV from latency.
Non-epigenetic mechanisms involve nuclear restriction of critical cellular transcription factors such as Nuclear factor-kappa Beta (NF-kB) or Nuclear factor of activated T-cells (NFAT) which activate transcription from the viral LTR, limiting nuclear levels of viral transcription transactivator protein Tat and its cellular co-factor; positive transcription elongation factor b (P-TEFb) which together regulate HIV transcriptional elongation.
The T-cell receptor (TCR) activation efficiently induces NF-kB, NFAT, and activator protein 1 (AP-1) transcription factors through multiple signal pathways and how these factors efficiently regulate HIV LTR transcription through the non-epigenetic mechanism.
Elongation factor P-TEFb induced through an extracellular signal-regulated kinase (ERK) dependent mechanism regulates HIV transcriptional elongation before Tat is synthesized and the role of AP-1 in the modulation of HIV transcriptional elongation through functional synergy with NF-kB.
The TCR signaling induces critical posttranslational modifications of the Cyclin-dependent kinase 9 (CDK9) subunit of P-TEFb which enhances interactions between P-TEFb and viral Tat protein and the resultant enhancement of HIV transcriptional elongation.
- T-cell receptor signaling
- HIV,
- Transcription
1. Introduction
With the introduction of the highly active antiretroviral therapy (HAART), the morbidity and mortality rate among HIV-infected individuals has reduced dramatically [1]. HAART, by virtue of restricting human immunodeficiency virus (HIV) levels, greatly enhances the host immune system [2]. The continuous use of HAART reduces the viremia in plasma beyond detection levels [3][4][5]. Nevertheless, using sensitive methods, residual viremia may still be detected in some individuals, validating the inability of HAART in eradicating HIV [6]. The ability of HIV to establish latent infections at the level of individual T-cells remains the main barrier to eradicate HIV [7]. The latent reservoir begins to establish quickly, within a few days of the initial infection [8]. The most-stable reservoir of HIV resides in the transcriptionally-silent resting memory CD4+ T-cells [9][10]. In order to express its genes, HIV requires host cell transcription machinery. Therefore, in transcriptionally-inert resting memory T-cells, HIV latency is established due to the unavailability of transcription factors [11]. Although the amount of latently infected cells is very limited (approximately one in one million of resting T-cells), a highly stable pool of latently-infected cells are always present in HIV patients, which remains a barrier for human immunodeficiency virus type-1 (HIV-1) eradication in patients undergoing effective HAART therapy [12]. The latent reservoir maintains its capacity to produce an infectious virus upon the discontinuation of the antiretroviral therapy (ART) and reactivation of harboring cells by recall antigens or various cytokines [13][14].
2. The T-Cell Receptor Signalosome
The T-cell receptor (TCR) is a multi-complex structure comprised of polymorphic protein molecules on the cell surface that interact with the peptide-MHC complex and non-polymorphic transmembrane CD3-zeta molecules, which transmit signals resulting from the TCR engagement and antigen recognition leading to T-cell activation [15][16]. The immediate outcome of the TCR/CD3 ligation by the peptide-MHC complex is the rapid phosphorylation of immunoreceptor tyrosine-based activation motifs (ITAMS) on the cytoplasmic tail of the TCR/CD3 complex by the Src family protein tyrosine kinase called leukocyte kinase-56 (Lck56) [15][17]. The phosphorylation of ITAMS leads to the recruitment of cytosolic tyrosine kinase, zeta-associated protein-70 (ZAP-70) to the plasma membrane where it phosphorylates multiple transmembrane adapter molecules including the T-lymphocyte-specific linker for activated T-cells (LAT) at multiple tyrosine residues [15][17][18]. Phosphorylated LAT recruits several multi-protein scaffold molecules including Grb-2, phospholipase C-gamma (PLC-g), and phosphoinositide-3 kinase (PI3K (Figure 1)).
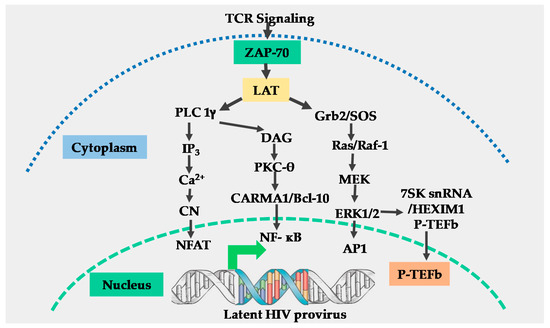
Figure 1. Schematic model for T-cell receptor signaling. Engagement of the T-cell receptor (TCR)/CD3 complex by the peptide-MHC complex requires the stabilization of the TCR-peptide/MHC complex interaction by CD4 or CD8 molecules, resulting in the activation of the p56 leukocyte kinase (Lck) which phosphorylates the immunoreceptor tyrosine-based activation motifs (ITAMS) within the cytoplasmic tails of CD3-zeta (not shown), resulting in the generation and transmission of the TCR signals. Engagement of the CD28 co-receptor (not shown) present on the surface of T-cells by B7 ligands on antigen-presenting cells is required for full TCR activation. Following full TCR activation, the zeta associated protein 70 (ZAP-70) and linker of activated T-cells (LAT) are activated. Activation of ZAP-70 and LAT results in the induction of three main signal pathways, namely: the calcium–calcineurin pathway which induces nuclear factor of activated T-cells (NFAT); the protein kinase C pathway which induces nuclear factor-kappa beta (NF-κB) and the MAP kinase/ extracellular signal-regulated kinase (ERK) pathway which induces activator protein 1 (AP-1) and positive transcription elongation factor b (P-TEFb). All these transcription factors translocate into the nucleus to regulate human immunodeficiency virus (HIV) transcription.
Grb-2 interacts with guanine nucleotide exchange factor SOS to activate the Ras-mitogen-activated protein kinase (MAPK) signal transduction pathway which leads to the induction of activator protein-1 (AP-1) [19] and P-TEFb through an ERK-dependent mechanism [20]. The hydrolysis of phosphatidylinositol 4, 5 bisphosphates (PIP-2) by PLC-gamma generates two-second messenger molecules; inositol 1, 4, 5 triphosphate (IP3) and diacylglycerol (DAG). The interaction of IP3 with its receptor at the cell membrane leads to intracellular Ca2+ mobilization resulting in activation of the calcium–calcineurin signal pathway that induces NFAT transcription factors (Figure 1) [15][18].
Through the canonical pathway of NF-κB induction, DAG activates the protein kinase C (PKC) pathway resulting in NF-κB mobilization via the CARMA1, Bcl110, and MALT1 (CBM) protein complex present upstream of the IκB-α kinase (IKK) complex [15][17][18]. Phosphatidylinositide-3 kinase enhances PKC-mediated NF-κB induction by activating Akt kinase which synergizes with the PKC activation pathway [21][22]. The efficient activation of the TCR/CD3 complex by the peptide-MHC complex ligation requires a co-stimulatory signal. Several families of co-stimulatory proteins are present on antigen-presenting cells (APCs) that interact with their receptors within the immunological synapse during the peptide-MHC presentation to T-lymphocytes [16][22]. The TCR-mediated antigen-specific stimulation is essential for initiating T-cell activation, signaling through the TCR has to be regulated through co-stimulatory or co-inhibitory receptors, which are important for full TCR activation or suppression of the T-cell responses [23]. The most important and well-studied co-stimulatory signal required for full TCR/CD3 activation is the B7 on APCs which, comprises CD80 and CD86 molecules. B7 interacts with CD28 molecules which are constitutively expressed on T-lymphocytes to enhance NF-κB induction resulting from TCR activation signals [16][22][24]. Other than the B7 co-stimulatory molecule, other co-stimulatory molecules that enhance the TCR signals have also been reported. For instance, Barat and Tremblay [25] demonstrated that the co-stimulation of CD43 along with the TCR activation augmented the nuclear mobilization of NF-κB and NFAT transcription factors independent of the CD28 co-stimulation. The augmented nuclear induction of NF-κB and NFAT enhanced HIV LTR transcription. In a different set of experiments, Tardif and Tremblay [26] demonstrated that CD81 also provides a co-stimulatory signal to the TCR signals which increased HIV LTR gene expression. On the other hand, the CTLA-4 and PD-1 are known to mediate the inhibitory functions in T-cell activation and signaling, and both are expressed only after cellular activation where they function in the feedback inhibition of T-cell activation. The PD-1 inhibits TCR activation by inducing the dephosphorylation of the TCR upstream signaling molecules through the transient recruitment of the phosphatase SHP2 while CTLA-4 competes with CD28 for CD80 and CD86 binding [27].
It is imperative to note that as much as independent signaling pathways usually lead to the induction of specific transcription factors, there is significant crosstalk between the TCR signaling pathways especially upstream of the kinases and phosphatases that activate the specific transcription factors. For instance, the LcK56, which is required for canonical TCR signaling leading to T-cell responses, also mediates a negative feedback loop through the phosphatase SHP2 that turns off the TCR signaling [28]. On the other hand, the HIV-1 protein nef has been reported to modulate the TCR activation signals to promote HIV replication and pathogenesis. For instance, Fenard et al. [29] reported that HIV nef increases the expression of the TCR-induced transcription factors NF-κB and NFAT following TCR activation. They reported that this was achieved through nef-induced priming of the TCR signaling pathways which occurs at a proximal step before protein kinase C (PKC) activation. Similarly, Neri et al. [30] also demonstrated that the modulation of TCR signaling by the HIV-1 nef results in a superinduction of NFAT transcription factor and interleukin 2 (IL-2) production. The superinduction of NFAT was shown to favor HIV-1 replication in both quiescent and metabolically active CD4 T-cells. These observations were confirmed by Fortin et al. [31]. Therefore, other than the inherent TCR modulation, the HIV nef protein can also modulate the TCR signaling pathways in order to enhance HIV transcription and pathogenesis.
References
- Quinn, T.C. HIV epidemiology and the effects of antiviral therapy on long-term consequences. AIDS 2008, 22 (Suppl. 3), S7–S12. [Google Scholar] [CrossRef]
- Pau, A.K.; George, J.M. Antiretroviral therapy: Current drugs. Infect. Dis. Clin. N. Am. 2014, 28, 371–402. [Google Scholar] [CrossRef]
- Persaud, D.; Siberry, G.K.; Ahonkhai, A.; Kajdas, J.; Monie, D.; Hutton, N.; Watson, D.C.; Quinn, T.C.; Ray, S.C.; Siliciano, R.F. Continued production of drug-sensitive human immunodeficiency virus type 1 in children on combination antiretroviral therapy who have undetectable viral loads. J. Virol. 2004, 78, 968–979. [Google Scholar] [CrossRef] [PubMed]
- Geeraert, L.; Kraus, G.; Pomerantz, R.J. Hide-and-seek: The challenge of viral persistence in HIV-1 infection. Annu. Rev. Med. 2008, 59, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Dornadula, G.; Zhang, H.; VanUitert, B.; Stern, J.; Livornese, L., Jr.; Ingerman, M.J.; Witek, J.; Kedanis, R.J.; Natkin, J.; DeSimone, J.; et al. Residual HIV-1 RNA in blood plasma of patients taking suppressive highly active antiretroviral therapy. JAMA 1999, 282, 1627–1632. [Google Scholar] [CrossRef] [PubMed]
- Sahu, G.K. Potential implication of residual viremia in patients on effective antiretroviral therapy. AIDS Res. Hum. Retrovir. 2015, 31, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Hokello, J.; Sharma, A.L.; Dimri, M.; Tyagi, M. Insights into the HIV Latency and the Role of Cytokines. Pathogens 2019, 8, 137. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ramratnam, B.; Tenner-Racz, K.; He, Y.; Vesanen, M.; Lewin, S.; Talal, A.; Racz, P.; Perelson, A.S.; Korber, B.T.; et al. Quantifying residual HIV-1 replication in patients receiving combination antiretroviral therapy. N. Engl. J. Med. 1999, 340, 1605–1613. [Google Scholar] [CrossRef]
- Vanhamel, J.; Bruggemans, A.; Debyser, Z. Establishment of latent HIV-1 reservoirs: What do we really know? J. Virus Erad. 2019, 5, 3–9. [Google Scholar] [CrossRef]
- Tyagi, M.; Pearson, R.J.; Karn, J. Establishment of HIV latency in primary CD4+ cells is due to epigenetic transcriptional silencing and P-TEFb restriction. J. Virol. 2010, 84, 6425–6437. [Google Scholar] [CrossRef]
- Tyagi, M.; Bukrinsky, M. Human immunodeficiency virus (HIV) latency: The major hurdle in HIV eradication. Mol. Med. 2012, 18, 1096–1108. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.J.; Siliciano, R.F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Ruelas, D.S.; Greene, W.C. An integrated overview of HIV-1 latency. Cell 2013, 155, 519–529. [Google Scholar] [CrossRef]
- Tyagi, M.; Iordanskiy, S.; Ammosova, T.; Kumari, N.; Smith, K.; Breuer, D.; Ilatovskiy, A.V.; Kont, Y.S.; Ivanov, A.; Uren, A.; et al. Reactivation of latent HIV-1 provirus via targeting protein phosphatase-1. Retrovirology 2015, 12, 63. [Google Scholar] [CrossRef] [PubMed]
- Kabouridis, P.S. Lipid rafts in T cell receptor signalling. Mol. Membr. Biol. 2006, 23, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Tseng, S.Y.; Dustin, M.L. T-cell activation: A multidimensional signaling network. Curr. Opin. Cell Biol. 2002, 14, 575–580. [Google Scholar] [CrossRef]
- Harder, T. Lipid raft domains and protein networks in T-cell receptor signal transduction. Curr. Opin. Immunol. 2004, 16, 353–359. [Google Scholar] [CrossRef]
- Zhang, J.T.; Han, E.; Liu, Y. Role of the ribosome in sequence-specific regulation of membrane targeting and translocation of P-glycoprotein signal-anchor transmembrane segments. J. Cell Sci. 2000, 113 Pt 14, 2545–2555. [Google Scholar]
- Zhang, W.; Samelson, L.E. The role of membrane-associated adaptors in T cell receptor signalling. Semin. Immunol. 2000, 12, 35–41. [Google Scholar] [CrossRef]
- Kim, Y.K.; Mbonye, U.; Hokello, J.; Karn, J. T-cell receptor signaling enhances transcriptional elongation from latent HIV proviruses by activating P-TEFb through an ERK-dependent pathway. J. Mol. Biol. 2011, 410, 896–916. [Google Scholar] [CrossRef]
- Kane, L.P.; Shapiro, V.S.; Stokoe, D.; Weiss, A. Induction of NF-kappaB by the Akt/PKB kinase. Curr. Biol. CB 1999, 9, 601–604. [Google Scholar] [CrossRef]
- Narayan, P.; Holt, B.; Tosti, R.; Kane, L.P. CARMA1 is required for Akt-mediated NF-kappaB activation in T cells. Mol. Cell. Biol. 2006, 26, 2327–2336. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Abe, R. Signal Transduction Via Co-stimulatory and Co-inhibitory Receptors. Adv. Exp. Med. Biol. 2019, 1189, 85–133. [Google Scholar] [CrossRef] [PubMed]
- Harhaj, E.W.; Maggirwar, S.B.; Good, L.; Sun, S.C. CD28 mediates a potent costimulatory signal for rapid degradation of IkappaBbeta which is associated with accelerated activation of various NF-kappaB/Rel heterodimers. Mol. Cell. Biol. 1996, 16, 6736–6743. [Google Scholar] [CrossRef]
- Barat, C.; Tremblay, M.J. Engagement of CD43 enhances human immunodeficiency virus type 1 transcriptional activity and virus production that is induced upon TCR/CD3 stimulation. J. Biol. Chem. 2002, 277, 28714–28724. [Google Scholar] [CrossRef]
- Tardif, M.R.; Tremblay, M.J. Tetraspanin CD81 provides a costimulatory signal resulting in increased human immunodeficiency virus type 1 gene expression in primary CD4+ T lymphocytes through NF-kappaB, NFAT, and AP-1 transduction pathways. J. Virol. 2005, 79, 4316–4328. [Google Scholar] [CrossRef]
- Saito, T. Molecular Dynamics of Co-signal Molecules in T-Cell Activation. Adv. Exp. Med. Biol. 2019, 1189, 135–152. [Google Scholar] [CrossRef]
- Ormonde, J.V.S.; Li, Z.; Stegen, C.; Madrenas, J. TAOK3 Regulates Canonical TCR Signaling by Preventing Early SHP-1-Mediated Inactivation of LCK. J. Immunol. 2018, 201, 3431–3442. [Google Scholar] [CrossRef]
- Fenard, D.; Yonemoto, W.; de Noronha, C.; Cavrois, M.; Williams, S.A.; Greene, W.C. Nef is physically recruited into the immunological synapse and potentiates T cell activation early after TCR engagement. J. Immunol. 2005, 175, 6050–6057. [Google Scholar] [CrossRef]
- Neri, F.; Giolo, G.; Potesta, M.; Petrini, S.; Doria, M. The HIV-1 Nef protein has a dual role in T cell receptor signaling in infected CD4+ T lymphocytes. Virology 2011, 410, 316–326. [Google Scholar] [CrossRef]
- Fortin, J.F.; Barat, C.; Beausejour, Y.; Barbeau, B.; Tremblay, M.J. Hyper-responsiveness to stimulation of human immunodeficiency virus-infected CD4+ T cells requires Nef and Tat virus gene products and results from higher NFAT, NF-kappaB, and AP-1 induction. J. Biol. Chem. 2004, 279, 39520–39531. [Google Scholar] [CrossRef] [PubMed]