Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Rita Xu and Version 1 by Ine Strubbe.
PHARC (polyneuropathy, hearing loss, ataxia, retinitis pigmentosa (RP) and early-onset cataract) is an acronym for a rare, neurodegenerative disease caused by biallelic variants in the ABHD12 gene
- PHARC syndrome
- ABHD12
- polyneuropathy
- hearing loss
1. Introduction
PHARC (polyneuropathy, hearing loss, ataxia, retinitis pigmentosa (RP) and early-onset cataract) is an acronym for a rare, neurodegenerative disease caused by biallelic variants in the ABHD12 gene [1,2][1][2]. ABHD12 is located on chromosome 20 and encodes the α/β-hydrolase domain-containing protein 12 (ABHD12), which is highly expressed in the central nervous system (CNS) and plays a vital role in lipid metabolism. In vitro, ABHD12 inactivates the main endocannabinoid lipid transmitter 2-arachidonyl glycerol (2-AG), which acts on the cannabinoid receptors 1 and 2 (CB1 and CB2) by converting the 2-AG into the metabolites arachidonate and glycerol [3,4][3][4]. In vivo, ABHD12 serves as a lyso-phosphatidylserine (lyso-PS) lipase, which degrades lyso-PS that is biosynthesized by ABHD16A [5]. Disruption of ABHD12 in mice leads to (i) accumulation of lyso-PS in the cerebellum breaching the homeostatic threshold, inducing continuous stimulation of the Purkinje neurons, leading to deregulated cerebellar activity and (ii) increased levels of microglial activation and inflammation [5,6,7][5][6][7]. Accompanying this inflammatory response in mice are behavioral deficits, including sensorimotor defects and hearing loss, which resembles the phenotype described in patients with PHARC syndrome [5,6,7][5][6][7].
Patients with PHARC syndrome demonstrate clinical variability with regard to disease onset, severity and progression [1,8,9,10][1][8][9][10]. Polyneuropathy is typically one of the first findings in patients with PHARC syndrome, which usually manifests in childhood. Early signs of polyneuropathy include distal muscle weakness, sensory disturbances, pes cavus and Achilles tendon contractures [1,9,11][1][9][11]. Sensorineural hearing loss is present in most patients with PHARC, with severity varying from moderate hearing loss to profound deafness [1,8][1][8]. RP is reported in the second or third decade of life, with fundoscopy showing optic disc pallor, retinal vessel attenuation and intraretinal specular hyperpigmentation [1,8][1][8]. As a result of RP, patients experience night blindness, constricted visual fields and, ultimately, central vision loss when retinal degeneration reaches the fovea [12]. While PHARC syndrome encompasses neurological, auditory and ophthalmic findings, not all of these findings are necessarily present at initial presentation [1,2][1][2]. Depending on the presenting symptoms, patients may first be misdiagnosed with other neurodegenerative diseases that give rise to roughly similar phenotypes, such as Charcot–Marie–Tooth, Usher type 3 and adult Refsum disease [1,11][1][11].
2. Patient Characteristics
A total of 15 patients from 12 different families were included in this study. An overview of the clinical and genetic characteristics of included patients is provided in Table 1. Most patients were male (n = 12; 80%) and the mean age at the most recent examination was 36.7 years (SD ± 11.0; range from 17.5 to 53.9). Previous (mis)diagnoses, available for 13 patients (87%), included forms of retinal degeneration (e.g., non-syndromic RP or Usher, n = 9; 69%), Charcot–Marie–Tooth (n = 2; 17%), spinocerebellar ataxia (n = 1; 8%) and optic neuropathy (n = 1; 8%). Phytanic acid levels were also assessed in 5 patients (A-1, B-2, C-3, C-4 and C-5) to rule out adult Refsum disease.
Table 1). Results from nerve conduction studies were available for nine patients (60%), which revealed various degrees of demyelinating polyneuropathy, even in an asymptomatic patient. Patient C-3 had no subjective complaints of sensory or motor deficits, despite both of his siblings (patients C-4 and C-5) being diagnosed with severe demyelinating polyneuropathy in childhood years. Still, upon neurological evaluation, a subtle foot drop and absent Achilles tendon reflexes were detected, with nerve conduction studies revealing a demyelinating polyneuropathy. Hearing loss was not subjectively present in two patients (patients C-3 and H-10), although formal audiometric testing results were not available in these patients. Similarly, the presence of ataxia was observed in less than half of the cohort, although the absence of ataxia could not be excluded in five patients as neurological examination was not performed or data were not available. MRI was performed in six patients (40%; patients A-1, C-3, C-4, C-5, E-7, J-12 and J-13), with signs of cerebellar atrophy in one patient with ataxia (patient C-5) and two patients without ataxia (J-12 and J-13).
Table 1.
Genetic and clinical characteristics at last examination of patients with biallelic
ABHD12
variants.
| Family-ID | Sex, Age | Genetic Analysis | Presence of PHARC Syndrome Symptoms and Age at Symptom Onset/Diagnosis (Years) | |||||
|---|---|---|---|---|---|---|---|---|
The ophthalmic findings in this cohort at the last visit are described in Table 2. Loss of BCVA was observed in all patients (100%), with a mean BCVA of 1.1 logMAR (SD ± 0.9; range from 0.1 to 2.8), which is equivalent to approximately 20/250 Snellen acuity. Four patients (A-1, B-2, E-7 and J-13), who carried the variant c.337_338delGAinsTTT in either homozygous or compound heterozygous form, had relatively preserved BCVA (BCVA ≥ 20/40 Snellen in the better-seeing eye). In contrast, the remaining patients, despite being in a similar age range, had visual acuities that could be classified as low vision (BCVA < 20/70 Snellen in the better-seeing eye) or worse. Patients with preserved BCVA were not significantly younger than those with low vision (−4.6 years, p = 0.496; independent t-test).
Table 2. Summary of ophthalmic findings at the most recent examination in this cohort of patients with biallelic ABHD12 variants.
| Family-ID | Sex, Age | BCVA (OD; OS) | Lens Status; Age at First Surgery | ffERG | Fundus Findings | |||
|---|---|---|---|---|---|---|---|---|
| Allele 1/Allele 2 | Protein Change | Polyneuropathy | Hearing Loss | Ataxia | Retinitis Pigmentosa | Cataract | ||
| Macular Changes | Bone Spicules | Spectral-Domain Optical Coherence Tomography | Fundus Autofluorescence | |||||
|---|---|---|---|---|---|---|---|---|
| A-1 | M, 47 | c.337_338delGAinsTTT/ c.1075del |
p. (Asp113Phefs*15)/p. (Val359Phefs*27) | Pes cavus, hammertoes, distal sensory loss and absent tendon reflexes; age 8 | Yes; age 28 | Yes; age 8 | Asymptomatic, detected during electrophysiological testing at age 45 | Yes; age 36 |
| A-1 | M, 47 | 20/22; 20/22 | Pseudophakic; surgery at age 36 | RCD | |||||||||||||||||||
| B-2 | F, 32 | c.337_338delGAinsTTT/ c.337_338delGAlinsTTT |
p. (Asp113Phefs*15)/p. (Asp113Phefs*15) | Yes; childhood | Yes; age 17 | RPE alterationsYes; age 45 | Reduced visual acuity; age 32 | Posterior subcapsular cataract; age 32 | |||||||||||||||
| No | Degeneration of the outer retina with preservation of ELM and EZ at the (para)fovea | Central hypo-AF surrounded by a hyper-AF ring | C-3 * | M, 33 | c.337_338delGAinsTTT/c.423-1_425del | p. (Asp113Phefs*15)/p. (?) | Asymptomatic; but detected during examination at age 27 | No ¶ | |||||||||||||||
| C-3 | Yes; age 27 | Night blindness; age 14 | Sutural cataract; age 3 | ||||||||||||||||||||
| M, 33 | 20/200; 20/200 | Pseudophakic; surgery at age 26 | NA | C-4 * | M, 33 | c.337_338delGAinsTTT/c.423-1_425del | p. (Asp113Phefs*15)/p. (?) | Star-shaped cataract; age 4 | |||||||||||||||
| RPE alterations | No | Atrophy | Yes | Degeneration of the outer retina | NA | Distal muscle weakness and sensory loss; childhood | Yes; NA | Yes; age 27 | Night blindness; age 21 | ||||||||||||||
| C-4 | M, 33 | Sutural cataract; age 3 | |||||||||||||||||||||
| 20/125; 20/100 | Pseudophakic; surgery at age 21 | MR | Atrophy | Yes | Epiretinal membrane, degeneration of the outer retina, CME ODS at age 29, resolved at age 31 | Hypo-AF lesions in the midperiphery with hyper-AF changes in the central macula | C-5 * | ||||||||||||||||
| C-5 | M, 38 | c.337_338delGAinsTTT/c.423-1_425del | p. (Asp113Phefs*15)/p. (?) | Abnormal gait pattern; childhood | Yes, 20 | Yes; age 31 | Night blindness | M, 38 | 20/134; 20/134 | Pseudophakic; surgery at age 19 | MR | Atrophy | Yes | Epiretinal membrane, degeneration of the outer retina, CME ODS at age 30, resolved at age 32 | NA | D-6 | M, 42 | c.477G > A/c.557G > C | p. (Trp159*)/p. (Arg186Pro) | Distal sensory loss and reduced tendon reflexes; age 35 | Yes; age 36 | Yes; NA | Reduced visual acuity; age 29 |
| D-6 | M, 42 | 20/400; 20/400 | Cortical cataract | RCD | Atrophy | No | Cortical cataract; age 29 | ||||||||||||||||
| Degeneration of the outer retina | E-7 | † | F, 36 | c.337_338delGAinsTTT/ c.337_338delGAlinsTTT |
p. (Asp113Phefs*15)/p. (Asp113Phefs*15) | NA | ‡ | Yes; age 12 | Yes; NA | Visual field loss; age 31 | Posterior subcapsular cataract; age 32 | ||||||||||||
| Central hypo-AF with a hyper-AF foveal spot | |||||||||||||||||||||||
| E-7 | F, 36 | 20/29; 20/29 | Pseudophakic; surgery at age 29 | RCD | RPE alterations | No | Degeneration of the outer retina with preservation of ELM and EZ at the (para)fovea | Hypo-AF regions in midperiphery with a macular hyper-AF ring | F-8 | M, 53 | c.784C > T/c.867 + 5G > A | p. (Arg262*)/ p. (?) |
Distal sensory loss; age 53 | ‡ | Yes; age 20 | NA | Reduced visual acuity; age 18 | No | |||||
| F-8 | M, 53 | LP; LP | Clear lens | NA | Atrophy | Yes | Extensive atrophy of all retinal layers | Generalized hypo-AF | G-9 | M, 34 | c.620-2A > G/c.620-2A > G | p. (?)/p. (?) | Lower limb muscle weakness; age 31 | ‡ | |||||||||
| G-9 | M, 34 | 20/400; 20/400 | Yes; age 20 | Pseudophakic; surgery at age 34 | RCD | AtrophyNA | Reduced visual acuity and night blindness; age 22 | Yes; age 26 | |||||||||||||||
| No | Extensive atrophy of all retinal layers at the fovea, with relatively preserved layers in the perifovea | Central hypo-AF | H-10 | † | M, 22 | c.193C > T/c.193 C > T | p. (Arg65*)/p. (Arg65*) | Lack of coordination; age 7 | |||||||||||||||
| H-10 | M, 22 | ‡ | No ¶ | NA | Reduced visual acuity; age 16 | No | |||||||||||||||||
| 20/240; 20/240 | Clear lens | RCD | Bull’s eye | No | Degeneration of the outer retina | Central hypo-AF | I-11 | † | M, 53 | c.374C > T/c.1154T > C | p. (Thr125Met)/p. (Leu385Pro) | NA, but epilepsy and learning difficulties | ‡ | Yes; age 44 | NA | Reduced visual acuity and night blindness; age 30 | Posterior polar cataract; age 41 | ||||||
| I-11 | J-12 * | M, 20 | c.337_338delGAinsTTT/c.337_338delGAinsTTT | p. (Asp113Phefs*15)/p. (Asp113Phefs*15) | c.337_338delGAinsTTT/c.337_338delGAinsTTT | p. (Asp113Phefs*15)/p. (Asp113Phefs*15) | Yes; age 18 | Yes; age 10 | No | Reduced visual acuity; age 10 | Star-shaped cataract; age 10 | ||||||||||||
| Epiretinal membrane, degeneration of the outer retina with preservation of ELM and EZ at the (para)fovea | Hypo-AF regions in midperiphery with a macular hyper-AF ring | ||||||||||||||||||||||
| B-2 | F, 32 | 20/25; 20/25 | Pseudophakic; surgery at age 32 | RCDM, 53 | HM; HM | Pseudophakic; surgery at age 44 | NA | Atrophy and Macular hole OS | Yes | Degeneration of the outer retina. Macular hole OS. | Mottled patches of hypo-AF in nasal region with hypo-AF in the central macula | Yes; age 20 | Yes; age 16 | No | |||||||||
| J-12 | Night blindness; age 16 | M, 20 | Star-shaped cataract; age 17 | ||||||||||||||||||||
| 20/200; 20/200 | Pseudophakic; surgery at age 20 | NA | Atrophy | No | Degeneration of the outer retina. | Central hypo-AF with hyper-AF borders | J-13 * | M, 17 | K-14 | F, 46 | c.1063C > T/c.1063C > T | p. (Arg355*)/p. (Arg355*) | |||||||||||
| J- 13 | Yes; age 47 | Yes; NA | Yes; NA | Yes; NA | Cerulean cataract, NA | ||||||||||||||||||
| M, 17 | L-15 | M, 39 | c.337_338delGAinsTTT/c.341dup | p. (Asp113Phefs*15)/p. (Leu114Phefs*14) | NA | ‡ | Yes; age 33 | NA | Night blindness; age 23 | Star-shaped cataract; age 29 |
Nucleotide numbering is based on reference sequence NM_001042472.3. * Patients C-3, C-4 and C-5 are siblings and patients J-12 and J-13 are siblings. † Consanguineous parents. ‡ Neurological evaluation/electrophysiological testing was not performed or available in these patients. ¶ Patients reported no subjective hearing loss, although audiometric testing results were not available. F = female; M = male; NA = data not available.
In total, 13 different ABHD12 variants were found in this cohort, 3 of which were missense variants, 3 splice site variants, 4 nonsense variants and 3 frameshift variants (Table 1 and Supplementary Table S1). The most common variant in this cohort was the frameshift variant c.337_338delGAinsTTT, which was present in more than half of the cohort in either homozygous or compound heterozygous form. This variant in exon 3 was predicted to result in a substitution of asparagine with phenylalanine at codon 113, introducing a premature termination codon (p.[Asp113Phefs*15]).
3. Clinical Examination
The onset of neurological, auditory and ophthalmic symptoms was variable, with no apparent order of symptom occurrence (
| 20/50; 20/40 |
| Star-shaped cataract |
| NA |
| Atrophy |
| No |
| Degeneration of the outer retina with preservation of ELM and EZ at the (para)fovea |
| Hyper-AF ring surrounded by a larger hyper-AF ring |
| K-14 | ||||||||
| F, 46 | ||||||||
| HM; 20/400 | ||||||||
| Cerulean cataract | ||||||||
| RCD | ||||||||
| Atrophy | ||||||||
| Yes | Degeneration of the outer retina. | Central hypo-AF with a hyper-AF foveal spot. Several hypo-AF lesions along the superior vascular arcade. | ||||||
| L-15 | M, 39 | 20/134; 20/200 | Pseudophakic; surgery at age 39 | RCD | Atrophy | Yes | Degeneration of the outer retina. CME ODS at age 33, resolved at age 39 | Generalized hypo-AF with preserved AF in the central macula. |
Slit-lamp examination revealed cataracts in 13 patients (87%), of whom 10 patients (77%) underwent uncomplicated cataract extraction. Various types of cataract were observed in this cohort, which also included congenital forms of cataract (Table 1). In four patients (patients C-5, J-12, J-13 and L-15), lens opacities were located in the posterior surface of the lens and followed a star-shaped distribution (Figure 1). Patients underwent their first cataract extraction and intraocular lens implementation at a mean age of 30.3 (SD ± 8.8; range from 19.0 to 44.0).
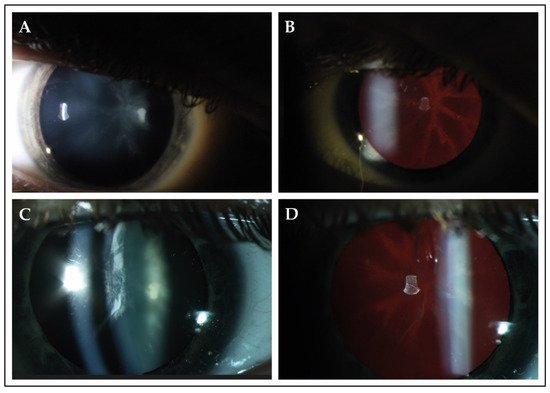
Figure 1. Slit-lamp findings in 2 patients with PHARC syndrome. (A,B) Slit-lamp photographs of the right eye of patient J-13 at the age of 17. Best-corrected visual acuity was 20/50 Snellen in this eye. Direct illumination demonstrated the presence of cataract in the posterior surface of the lens. Retroillumination revealed that the observed opacity followed a star-shaped distribution, which seemed to delineate the crystalline lens sutures of the posterior cortex. (C,D) The right eye of patient L-15 (age 37) showed opacities in both the anterior and posterior cortex. Best-corrected visual acuity was 20/100 during this visit. Retroillumination showed anterior cortical cataract and a star-shaped opacity in the posterior surface.
In Figure 2, we present representative fundus and multimodal imaging findings of this cohort. Fundus examination revealed signs of retinal degeneration in all patients, although a clinical hallmark of RP—intraretinal spicular hyperpigmentation—was only observed in 7 out of 15 patients (47%; Table 2). Patients with intraretinal spicular hyperpigmentation had worse logMAR BCVA than those without pigmentation (+0.9 logMAR BCVA, p = 0.019; independent t-test). Macular involvement was present in all patients (100%), ranging from retinal pigment epithelium alterations to macular atrophy. Full-field electroretinography data were available for 10 patients (67%), showing a rod-cone dystrophy pattern (n = 8; 80%) or minimal scotopic and photopic responses (n = 2; 20%) (Supplemental Figure S1).
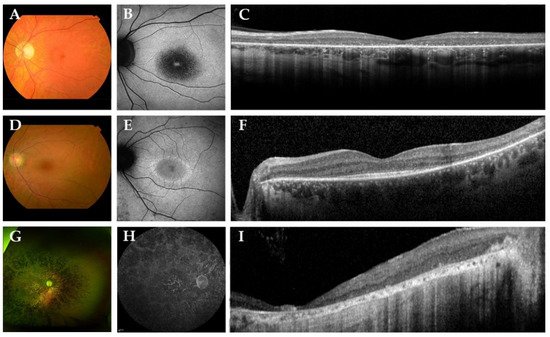
Figure 2. Representative color fundus photographs with corresponding fundus autofluorescence (FAF) and spectral-domain optical coherence tomography (SD-OCT) images in this cohort of patients with biallelic ABHD12 variants. (A–C) The left eye of patient D-6, a 42-year-old man with Snellen best-corrected visual acuity (BCVA) of 20/400. Fundus photography revealed a slightly pale optic disc, atrophic macular changes and retinal pigment epithelium (RPE) changes in the midperipheral retina, in the absence of spicular hyperpigmentation. FAF imaging showed a region of hypo-autofluorescence (hypo-AF) in the central macula with a hyper-autofluorescent (hyper-AF) spot in the fovea. On SD-OCT, loss of the external limiting membrane and ellipsoid zone was observed. (D–F) The left eye of a 36-year-old woman, patient E-7, with Snellen BCVA of 20/29. Fundus imaging showed macular and midperipheral alterations, with no evident spicular hyperpigmentation. On FAF imaging, hypo-AF zones of RPE degeneration were present outside the macula and a macular hyper-AF ring was observed. SD-OCT showed preservation of the outer retinal layers in the (para)fovea. (G–I) Patient F-8, a 53-year-old man with light perception visual acuity, showed extensive degeneration across the entire retina with dense spicular hyperpigmentation reaching the posterior pole. FAF imaging demonstrated generalized hypo-AF due to the extensive RPE atrophy. SD-OCT showed marked chorioretinal atrophy.
References
- Fiskerstrand, T.; H’Mida-Ben Brahim, D.; Johansson, S.; M’Zahem, A.; Haukanes, B.I.; Drouot, N.; Zimmermann, J.; Cole, A.J.; Vedeler, C.; Bredrup, C.; et al. Mutations in ABHD12 Cause the Neurodegenerative Disease PHARC: An Inborn Error of Endocannabinoid Metabolism. Am. J. Hum. Genet. 2010, 87, 410–417.
- Fiskerstrand, T.; Knappskog, P.; Majewski, J.; Wanders, R.J.; Boman, H.; Bindoff, L.A. A novel Refsum-like disorder that maps to chromosome 20. Neurology 2009, 72, 20.
- Savinainen, J.R.; Saario, S.M.; Laitinen, J.T. The serine hydrolases MAGL, ABHD6 and ABHD12 as guardians of 2-arachidonoylglycerol signalling through cannabinoid receptors. Acta Physiol. (Oxf.) 2012, 204, 267–276.
- Blankman, J.L.; Simon, G.M.; Cravatt, B.F. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem. Biol. 2007, 14, 1347–1356.
- Shin, M.; Ware, T.B.; Lee, H.-C.; Hsu, K.-L. Lipid-metabolizing serine hydrolases in the mammalian central nervous system: Endocannabinoids and beyond. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 907–921.
- Joshi, A.; Shaikh, M.; Singh, S.; Rajendran, A.; Mhetre, A.; Kamat, S.S. Biochemical characterization of the PHARC-associated serine hydrolase ABHD12 reveals its preference for very-long-chain lipids. J. Biol. Chem. 2018, 293, 16953–16963.
- Kamat, S.S.; Camara, K.; Parsons, W.H.; Chen, D.-H.; Dix, M.M.; Bird, T.D.; Howell, A.R.; Cravatt, B.F. Immunomodulatory lysophosphatidylserines are regulated by ABHD16A and ABHD12 interplay. Nat. Chem. Biol. 2015, 11, 164–171.
- Chen, D.-H.; Naydenov, A.; Blankman, J.L.; Mefford, H.C.; Davis, M.; Sul, Y.; Barloon, A.S.; Bonkowski, E.; Wolff, J.; Matsushita, M.; et al. Two novel mutations in ABHD12: Expansion of the mutation spectrum in PHARC and assessment of their functional effects. Hum. Mutat. 2013, 34, 1672–1678.
- Eisenberger, T.; Slim, R.; Mansour, A.; Nauck, M.; Nürnberg, G.; Nürnberg, P.; Decker, C.; Dafinger, C.; Ebermann, I.; Bergmann, C.; et al. Targeted next-generation sequencing identifies a homozygous nonsense mutation in ABHD12, the gene underlying PHARC, in a family clinically diagnosed with Usher syndrome type 3. Orphanet J. Rare Dis. 2012, 7, 59.
- Nishiguchi, K.M.; Avila-Fernandez, A.; van Huet, R.A.C.; Corton, M.; Pérez-Carro, R.; Martín-Garrido, E.; López-Molina, M.I.; Blanco-Kelly, F.; Hoefsloot, L.H.; van Zelst-Stams, W.A.; et al. Exome Sequencing Extends the Phenotypic Spectrum for ABHD12 Mutations: From Syndromic to Nonsyndromic Retinal Degeneration. Ophthalmology 2014, 121, 1620–1627.
- Thimm, A.; Rahal, A.; Schoen, U.; Abicht, A.; Klebe, S.; Kleinschnitz, C.; Hagenacker, T.; Stettner, M. Genotype-phenotype correlation in a novel ABHD12 mutation underlying PHARC syndrome. J. Peripher. Nervous Syst. 2020, 25, 112–116.
- Verbakel, S.K.; van Huet, R.A.C.; Boon, C.J.F.; den Hollander, A.I.; Collin, R.W.J.; Klaver, C.C.W.; Hoyng, C.B.; Roepman, R.; Klevering, B.J. Non-syndromic retinitis pigmentosa. Prog. Retin. Eye Res. 2018, 66, 157–186.
More