Gene therapy serves as a promising therapy in the pipeline for treatment of epidermolysis bullosa (EB). However, with great promise, the risk of autoimmunity must be considered. While EB is a group of inherited blistering disorders caused by mutations in various skin proteins, autoimmune blistering diseases (AIBD) have a similar clinical phenotype and are caused by autoantibodies targeting skin antigens. Often, AIBD and EB have the same protein targeted through antibody or mutation, respectively. Moreover, EB patients are also reported to carry anti-skin antibodies of questionable pathogenicity. It has been speculated that activation of autoimmunity is both a consequence and cause of further skin deterioration in EB due to a state of chronic inflammation.
- gene therapy
- epidermolysis bullosa
- autoimmunity
- autoimmune blistering disorder
- collagen XVII
1. Introduction
| Antigen | EB Subtype | AIBD Subtype |
|---|---|---|
| BP230 (dystonin) | EBS | Bullous pemphigoid |
| Collagen XVII (BP180) | JEB | Bullous pemphigoid, Pemphigoid gestationis |
| Laminin 332 | JEB | Mucous membrane pemphigoid |
| α6β4 integrin | JEB | Mucous membrane pemphigoid |
| Collagen VII | DEB | Epidermolysis bullosa acquisita |
2. Presence of Anti-Skin Antibodies in EB
| EB Subtype | n | Autoantigen | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Collagen | FN | LAM | Dsg1 | Dsg3 | Collagen XVII/ BP180 | BP230 | |||||||||
| I | II | III | IV | V | VI | VII | |||||||||
| [14] | EBA | 2 | 0.0% | 0.0% | 100.0% | 0.0% | 100.0% | 0.0% | 0.0% | 50.0% | |||||
| EBS | 20 | 0.0% | 0.0% | 85.0% | IIF: IgG binding to the dermal side of the salt-split skin ELISA: 60.0% |
Positive for anti-collagen type VII, anti-BP180, and anti-BP230 Immunoblot Analysis: Reactive to laminin 33285.0% |
0.0% |
Mutations:0.0% | 40.0% |
||||||
| c.410G > A and c.3674C > T in the COL7A1 gene | JEB | 4 | 0.0% | 0.0% | |||||||||||
| 2019 | 100.0% | Fania | JEB | BP | DIF: Linear IgG and C3 deposits in an n-serrated pattern at the DEJ IIF: Epidermal staining of the salt-split skin ELISA: Positive for anti-BP180. Negative for anti-BP230 Immunoblot analysis: Reactive to BP180 and its LAD-1 domain. Not reactive to laminin 332 Mutations: 50.0% |
100.0% | 25.0% | 0.0% | c.1132 + 5G > A in the LAMB3 gene0.0% | ||||||
| DEB | 6 | 0.0% | 16.7% | 83.3% | 33.3% | 100.0% | 16.7% | 16.7% | 66.7% | ||||||
| Total | 32 | 0.0% | 3.1% | 87.5% | 50.0% | 90.6% | 6.3% | 3.1% | 40.6% | ||||||
| [15] | RDEB | 19 | 4.96 U/mL | 5.62 U/mL | 6.14 U/mL | 14.2 U/mL | 12.7 U/mL | ||||||||
| Other EB | 23 | 1.08 U/mL | 2.67 U/mL | 2.8 U/mL | 5.7 U/mL | 3.7 U/mL | |||||||||
| Healthy Controls | 38 | 0.26 U/mL | 2.12 U/mL | 1.58 U/mL | 1.82 U/mL | 1.68 U/mL | |||||||||
| [16] | RDEB | 17 | 88% | combined percentage of 88% | |||||||||||
| EBS | 10 | 10% | combined percentage of 50% | ||||||||||||
| Year | Author | EB Type | AIBD Type | Workup |
|---|---|---|---|---|
| 2016 | Hayashi | DDEB | EBA | DIF: Linear deposits of IgG and C3 at the DEJ IIF: Linear deposition of IgG at the dermal side of the DEJ Immunoblot analysis: Reactive to collagen type VII and its NC1 domain. Non-reactive to laminin 322 Mutations: c.7868G > A in the COL7A1 gene |
| 2018 | Guerra | RDEB | EBA | DIF: Linear deposition of IgG with a u-serrated pattern along the cutaneous BMZ |
3. Dysregulated Inflammatory Response and Blister Formation
When subjected to trauma, keratinocytes display increased sensitivity to autoantibodies [40]. Collagen XVII is a known inhibitor of keratinocyte migration, while its shed ectodomain leads to stabilization and cell immobilization [41]. In nonlethal JEB, the absence of collagen XVII or dysfunctional interaction between laminin-332 and collagen XVII is speculated to promote keratinocyte migration [41][42][43]. Thus, the weakened attachment of keratinocytes to the basement membrane, increased keratinocyte sensitivity to circulating autoantibodies, and subsequent expression of eosinophil chemotactic factors enhance deposition of antibodies and facilitate blister formation [44]. However, blister formation has a much more complex etiology where the interplay between cytokines, chemokines, and MMP is important. In patients with JEB, anti-collagen XVII autoantibodies trigger the release of inflammatory cytokines that may exacerbate DEJ separation [45]. In vitro, when JEB-derived (collagen-XVII-deficient) epidermal keratinocytes are exposed to inflammatory stimuli (ultraviolet B radiation, lipopolysaccharide, phorbol 12-myristate 13-acetate, and tumor necrosis factor), an abnormally high IL-8 response is seen [45]. As a chemotactic agent, IL-8 contributes to neutrophil recruitment [46]. In turn, re-induction of collagen XVII expression normalizes this response (against lipopolysaccharide and ultraviolet B radiation), suggesting that it may serve as a pathway for inflammation and subsequent lesion formation in the skin of collagen-XVII-deficient EB patients [45]. Although not fully elucidated, antibody-mediated disruption of interactions between collagen XVII and other components of the BM may result in a pro-inflammatory response. In the complement-independent pathway, the binding of autoantibody to collagen XVII and the subsequent internalization of these immune complexes causes a depletion of collagen XVII from cell surface [47]. This results in the formation of collagen XVII-deficient hemidesmosomes that weaken the adhesion in a patient’s skin [47]. Thus, BP-IgG may induce or exacerbate skin fragility by itself [28].4. Implications of Autoantibodies in Gene Therapy
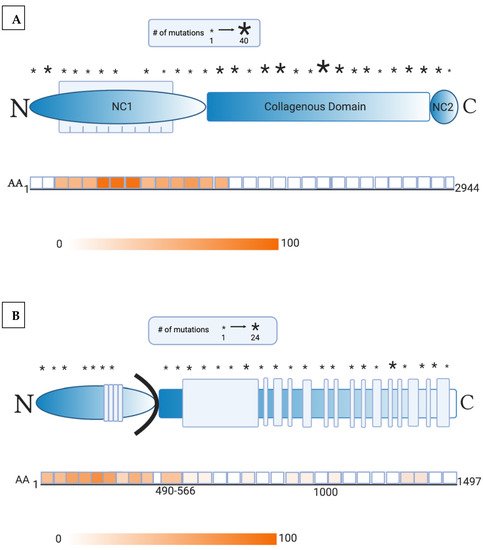
Recent advancements in the understanding of EB pathogenesis have allowed researchers to identify novel treatment options, including gene therapy. Initial success in gene therapy was uncovered for JEB patients with a LAMB3 mutation, who received genetically engineered epidermal sheet grafts overexpressing an ex vivo, retroviral full-length LAMB3 transgenic product [48][49]. LAMB3 expression was maintained within the holoclonal epidermal stem cells and laminin 332 was found in the DEJ until 21 months [49]. However, gene therapy-mediated expression of a functional protein runs the risk of inducing autoimmunity. Fortunately, these study patients did not generate an immune reaction to the antigenic laminin β3 chain [50], likely because the selected patients had missense mutations involving a single amino acid or small deletions [48][49]. On the other hand, JEB patients with null mutations and fatal disease would be expected to develop immunoreactivity, and laminin-332 expression in skin via gene therapy would not correct severe upper respiratory, kidney, or internal disease [3]. Notably, autoantibodies against laminin β3 are also uncommon in patients with laminin-332 pemphigoid, occurring in less than 30% of patients [51]. However, autoantibodies against laminin α3 are present in close to 90%. This suggests that LAMB3 may be a less immunogenic target, thus contributing to the success of LAMB3 correction. Gene therapy in recessive DEB patients is even more challenging due to the large size of COL7A1 and the increased immunogenicity of NC1 [52]. Epidermal sheet grafting maintained collagen VII expression in the primary collagen-VII-deficient recessive DEB keratinocytes of immunodeficient mice [53]. However, in order to avoid the risk of autoimmunity, human trials excluded patients without positive expression to the NC1 domain of collagen VII, the most antigenic portion of this protein [6]. While NC1 is a highly targeted epitope in EBA, only approximately 30% of patient cell cultures fail to express NC1 [54]. Another important criterion was selection of patients with severe generalized recessive DEB showing the absence of expression of full-length collagen VII (near the NC2 domain) [55]. All patients tolerated grafting with collagen-VII-engineered autologous epidermal sheets without adverse events and skin biopsy demonstrated linear collagen VII expression [55][56]. Clinical improvement in wound healing was more profound in grafted sites and patient-reported pain, itch, and wound durability [55]. Only one patient in this study developed autoantibodies (specific to NC2 domain of collagen VII), but pre-therapy serum Western blot analysis showed low levels of transient collagen VII antibodies despite an initial negative screening with indirect immunofluorescence [55]. It is likely that immunoreactivity pre-existed in this patient, and gene therapy exacerbated the immune response. Screening data revealed that this patient expressed a collagen VII molecule containing NC1 domain but not NC2, suggesting that this patient’s reaction was possibly an allo-reaction to the therapeutic gene product [55]. Thus, this patient did not experience increased blistering outside of the treated areas. Although low quantities of collagen VII antibodies in recessive DEB patients are considered nonpathogenic, as in this patient’s case [56][57][58], caution is advised when these patients are treated with gene therapy. Nonsense mutations, prevalent in 30% of recessive DEB patients, result in a premature termination codon that generates a truncated collagen VII product. In a recent trial of these recessive DEB patients treated with intravenous gentamicin, a premature termination codon readthrough was induced which created a new type VII collagen and anchoring fibrils that persisted for 3 months. Preliminary results demonstrate that none of the patients developed autoantibodies to collagen VII despite aminoglycoside-induced production of new collagen VII [59].References
- Patel, P.M.; Jones, V.A.; Murray, T.N.; Amber, K.T. A Review Comparing International Guidelines for the Management of Bullous Pemphigoid, Pemphigoid Gestationis, Mucous Membrane Pemphigoid, and Epidermolysis Bullosa Acquisita. Am. J. Clin. Dermatol. 2020, 21, 557–565.
- Jones, V.A.; Patel, P.M.; Amber, K.T. Eosinophils in Bullous Pemphigoid. Panminerva Med. 2020.
- Marinkovich, M.P.; Tang, J.Y. Gene Therapy for Epidermolysis Bullosa. J. Investig. Dermatol. 2019, 139, 1221–1226.
- Has, C.; Nyström, A.; Saeidian, A.H.; Bruckner-Tuderman, L.; Uitto, J. Epidermolysis bullosa: Molecular pathology of connective tissue components in the cutaneous basement membrane zone. Matrix Biol. 2018, 71, 313–329.
- Mayr, E.; Koller, U.; Bauer, J.W. Gene Therapy for the COL7A1 Gene. In Gene Therapy; Molina, F.M., Ed.; IntechOpen: Rijeka, Croatia, 2013.
- Lapiere, J.C.; Woodley, D.T.; Parente, M.G.; Iwasaki, T.; Wynn, K.C.; Christiano, A.M.; Uitto, J. Epitope mapping of type VII collagen. Identification of discrete peptide sequences recognized by sera from patients with acquired epidermolysis bullosa. J. Clin. Investig. 1993, 92, 1831–1839.
- Dang, N.; Murrell, D.F. Mutation analysis and characterization of COL7A1 mutations in dystrophic epidermolysis bullosa. Exp. Dermatol. 2008, 17, 553–568.
- Tanaka, T.; Furukawa, F.; Imamura, S. Epitope mapping for epidermolysis bullosa acquisita autoantibody by molecularly cloned cDNA for type VII collagen. J. Investig. Dermatol. 1994, 102, 706–709.
- Dresow, S.K.; Sitaru, C.; Recke, A.; Oostingh, G.J.; Zillikens, D.; Gibbs, B.F. IgE autoantibodies against the intracellular domain of BP180. Br. J. Dermatol. 2009, 160, 429–432.
- Di Zenzo, G.; Grosso, F.; Terracina, M.; Mariotti, F.; De Pità, O.; Owaribe, K.; Mastrogiacomo, A.; Sera, F.; Borradori, L.; Zambruno, G. Characterization of the anti-BP180 autoantibody reactivity profile and epitope mapping in bullous pemphigoid patients. J. Investig. Dermatol. 2004, 122, 103–110.
- Bauer, J.W.; Lanschuetzer, C. Type XVII collagen gene mutations in junctional epidermolysis bullosa and prospects for gene therapy. Clin. Exp. Dermatol. 2003, 28, 53–60.
- Condrat, I.; He, Y.; Cosgarea, R.; Has, C. Junctional Epidermolysis Bullosa: Allelic Heterogeneity and Mutation Stratification for Precision Medicine. Front. Med. 2018, 5, 363.
- Pasmooij, A.M.G.; Pas, H.H.; Jansen, G.H.L.; Lemmink, H.H.; Jonkman, M.F. Localized and generalized forms of blistering in junctional epidermolysis bullosa due to COL17A1 mutations in the Netherlands. Br. J. Dermatol. 2007, 156, 861–870.
- Gay, S.; Fine, J.D.; Storer, J.S. Autoantibodies to extracellular collagen matrix components in epidermolysis bullosa and other bullous diseases. Arch. Dermatol. Res. 1988, 280, 333–337.
- Esposito, S.; Guez, S.; Orenti, A.; Tadini, G.; Scuvera, G.; Corti, L.; Scala, A.; Biganzoli, E.; Berti, E.; Principi, N. Autoimmunity and Cytokine Imbalance in Inherited Epidermolysis Bullosa. Int. J. Mol. Sci. 2016, 17, 1625.
- Tampoia, M.; Bonamonte, D.; Filoni, A.; Garofalo, L.; Morgese, M.G.; Brunetti, L.; Di Giorgio, C.; Annicchiarico, G. Prevalence of specific anti-skin autoantibodies in a cohort of patients with inherited epidermolysis bullosa. Orphanet J. Rare Dis. 2013, 8, 132.
- Annicchiarico, G.; Morgese, M.G.; Esposito, S.; Lopalco, G.; Lattarulo, M.; Tampoia, M.; Bonamonte, D.; Brunetti, L.; Vitale, A.; Lapadula, G.; et al. Proinflammatory Cytokines and Antiskin Autoantibodies in Patients With Inherited Epidermolysis Bullosa. Medicine 2015, 94, e1528.
- Kasperkiewicz, M.; Hirose, M.; Recke, A.; Schmidt, E.; Zillikens, D.; Ludwig, R.J. Clearance rates of circulating and tissue-bound autoantibodies to type VII collagen in experimental epidermolysis bullosa acquisita. Br. J. Dermatol. 2010, 162, 1064–1070.
- Licarete, E.; Ganz, S.; Recknagel, M.J.; Di Zenzo, G.; Hashimoto, T.; Hertl, M.; Zambruno, G.; Hundorfean, G.; Mudter, J.; Neurath, M.F.; et al. Prevalence of collagen VII-specific autoantibodies in patients with autoimmune and inflammatory diseases. BMC Immunol. 2012, 13, 16.
- Hayashi, R.; Natsuga, K.; Watanabe, M.; Iwata, H.; Shinkuma, S.; Ito, A.; Masui, Y.; Ito, M.; Shimomura, Y. Epidermolysis Bullosa Acquisita Develops in Dominant Dystrophic Epidermolysis Bullosa. J. Investig. Dermatol. 2016, 136, 320–323.
- Gammon, W.R.; Briggaman, R.A.; Woodley, D.T.; Heald, P.W.; Wheeler, E.C., Jr. Epidermolysis bullosa acquisita—a pemphigoid-like disease. J. Am. Acad. Dermatol. 1984, 11, 820–832.
- Stevens, N.E.; Cowin, A.J.; Kopecki, Z. Skin Barrier and Autoimmunity-Mechanisms and Novel Therapeutic Approaches for Autoimmune Blistering Diseases of the Skin. Front. Immunol. 2019, 10, 1089.
- Mihai, S.; Hirose, M.; Wang, Y.; Thurman, J.M.; Holers, V.M.; Morgan, B.P.; Kohl, J.; Zillikens, D.; Ludwig, R.J.; Nimmerjahn, F. Specific Inhibition of Complement Activation Significantly Ameliorates Autoimmune Blistering Disease in Mice. Front. Immunol. 2018, 9, 535.
- Mihai, S.; Chiriac, M.T.; Takahashi, K.; Thurman, J.M.; Holers, V.M.; Zillikens, D.; Botto, M.; Sitaru, C. The alternative pathway of complement activation is critical for blister induction in experimental epidermolysis bullosa acquisita. J. Immunol. 2007, 178, 6514–6521.
- Sitaru, C.; Kromminga, A.; Hashimoto, T.; Brocker, E.B.; Zillikens, D. Autoantibodies to type VII collagen mediate Fcgamma-dependent neutrophil activation and induce dermal-epidermal separation in cryosections of human skin. Am. J. Pathol. 2002, 161, 301–311.
- Bieber, K.; Witte, M.; Sun, S.; Hundt, J.E.; Kalies, K.; Drager, S.; Kasprick, A.; Twelkmeyer, T.; Manz, R.A.; Konig, P.; et al. T cells mediate autoantibody-induced cutaneous inflammation and blistering in epidermolysis bullosa acquisita. Sci. Rep. 2016, 6, 38357.
- Liu, Z.; Giudice, G.J.; Swartz, S.J.; Fairley, J.A.; Till, G.O.; Troy, J.L.; Diaz, L.A. The role of complement in experimental bullous pemphigoid. J. Clin. Investig. 1995, 95, 1539–1544.
- Iwata, H.; Ujiie, H. Complement-independent blistering mechanisms in bullous pemphigoid. Exp. Dermatol. 2017, 26, 1235–1239.
- Mihai, S.; Chiriac, M.T.; Herrero-González, J.E.; Goodall, M.; Jefferis, R.; Savage, C.O.S.; Zillikens, D.; Sitaru, C. IgG4 autoantibodies induce dermal-epidermal separation. J. Cell. Mol. Med. 2007, 11, 1117–1128.
- Liu, Y.; Li, L.; Xia, Y. BP180 Is Critical in the Autoimmunity of Bullous Pemphigoid. Front. Immunol. 2017, 8, 1752.
- Liu, Y.-D.; Wang, Y.-H.; Ye, Y.-C.; Zhao, W.-L.; Li, L. Prognostic factors for mortality in patients with bullous pemphigoid: A meta-analysis. Arch. Dermatol. Res. 2017, 309, 335–347.
- van Beek, N.; Lüttmann, N.; Huebner, F.; Recke, A.; Karl, I.; Schulze, F.S.; Zillikens, D.; Schmidt, E. Correlation of Serum Levels of IgE Autoantibodies Against BP180 With Bullous Pemphigoid Disease Activity. JAMA Dermatol. 2017, 153, 30–38.
- Wieland, C.N.; Comfere, N.I.; Gibson, L.E.; Weaver, A.L.; Krause, P.K.; Murray, J.A. Anti-bullous pemphigoid 180 and 230 antibodies in a sample of unaffected subjects. Arch Dermatol. 2010, 146, 21–25.
- Málaga, S.; Toral, J.F.; Santos, F.; Riesgo, I.; Crespo, M. Renal amyloidosis complicating a recessive epidermolysis bullosa in childhood. Helv. Paediatr. Acta 1983, 38, 167–170.
- Mann, J.F.; Zeier, M.; Zilow, E.; Schärer, K.; Anton-Lamprecht, I.; Waldherr, R.; Andrassy, K.; Ritz, E. The spectrum of renal involvement in epidermolysis bullosa dystrophica hereditaria: Report of two cases. Am. J. Kidney Dis. 1988, 11, 437–441.
- Chen, C.C.; Isomoto, H.; Hayashi, T. Gastrointestinal amyloidosis secondary to inherited skin disorder. Gastroenterology 2012, 142, e9–e10.
- Annicchiarico, G.; Morgese, M.G.; Brunetti, L.; Tampoia, M.; Garofalo, L.; Aceto, G.; Fiore, T.; Mauro, S.; Minelli, M. Improvement of renal function in epidermolysis bullosa patients after gluten free diet: Two cases. Eur. Rev. Med. Pharmacol. Sci. 2012, 16, 138–141.
- Guerra, L.; Condorelli, A.G.; Fortugno, P.; Calabresi, V.; Pedicelli, C.; Di Zenzo, G.; Castiglia, D. Epidermolysis Bullosa (EB) Acquisita in an Adult Patient with Previously Unrecognized Mild Dystrophic EB and Biallelic COL7A1 Mutations. Acta Derm. Venereol. 2018, 98, 411–415.
- Fania, L.; Provini, A.; Salemme, A.; Sinagra, J.L.; Guerra, L.; Mazzanti, C.; Didona, B.; Castiglia, D.; Di Zenzo, G. Development of bullous pemphigoid in junctional epidermolysis bullosa. J. Eur. Acad. Dermatol. Venereol. 2020, 34, e146–e148.
- Balighi, K.; Daneshpazhooh, M.; Azizpour, A.; Lajevardi, V.; Mohammadi, F.; Chams-Davatchi, C. Koebner phenomenon in pemphigus vulgaris patients. JAAD Case Rep. 2016, 2, 419–421.
- Tasanen, K.; Tunggal, L.; Chometon, G.; Bruckner-Tuderman, L.; Aumailley, M. Keratinocytes from patients lacking collagen XVII display a migratory phenotype. Am. J. Pathol. 2004, 164, 2027–2038.
- Jonkman, M.F.; de Jong, M.C.; Heeres, K.; Pas, H.H.; van der Meer, J.B.; Owaribe, K.; de Velasco, A.M.M.; Niessen, C.M.; Sonnenberg, A. 180-kD bullous pemphigoid antigen (BP180) is deficient in generalized atrophic benign epidermolysis bullosa. J. Clin. Investig. 1995, 95, 1345–1352.
- McGrath, J.A.; Gatalica, B.; Christiano, A.M.; Li, K.; Owaribe, K.; McMillan, J.R.; Eady, R.A.; Uitto, J. Mutations in the 180-kD bullous pemphigoid antigen (BPAG2), a hemidesmosomal transmembrane collagen (COL17A1), in generalized atrophic benign epidermolysis bullosa. Nat. Genet. 1995, 11, 83–86.
- Jones, V.A.; Patel, P.M.; Gibson, F.T.; Cordova, A.; Amber, K.T. The Role of Collagen XVII in Cancer: Squamous Cell Carcinoma and Beyond. Front. Oncol. 2020, 10, 352.
- Van den Bergh, F.; Eliason, S.L.; Burmeister, B.T.; Giudice, G.J. Collagen XVII (BP180) modulates keratinocyte expression of the proinflammatory chemokine, IL-8. Exp. Dermatol. 2012, 21, 605–611.
- Liu, Z.; Giudice, G.J.; Zhou, X.; Swartz, S.J.; Troy, J.L.; Fairley, J.A.; Till, G.O.; Diaz, L.A. A major role for neutrophils in experimental bullous pemphigoid. J. Clin. Investig. 1997, 100, 1256–1263.
- Iwata, H.; Kamaguchi, M.; Ujiie, H.; Nishimura, M.; Izumi, K.; Natsuga, K.; Shinkuma, S.; Nishie, W.; Shimizu, H. Macropinocytosis of type XVII collagen induced by bullous pemphigoid IgG is regulated via protein kinase C. Lab. Investig. 2016, 96, 1301–1310.
- Bauer, J.W.; Koller, J.; Murauer, E.M.; De Rosa, L.; Enzo, E.; Carulli, S.; Bondanza, S.; Recchia, A.; Muss, W.; Diem, A.; et al. Closure of a Large Chronic Wound through Transplantation of Gene-Corrected Epidermal Stem Cells. J. Investig. Dermatol. 2017, 137, 778–781.
- Hirsch, T.; Rothoeft, T.; Teig, N.; Bauer, J.W.; Pellegrini, G.; De Rosa, L.; Scaglione, D.; Reichelt, J.; Klausegger, A.; Kneisz, D.; et al. Regeneration of the entire human epidermis using transgenic stem cells. Nature 2017, 551, 327–332.
- Domloge-Hultsch, N.; Gammon, W.R.; Briggaman, R.A.; Gil, S.G.; Carter, W.G.; Yancey, K.B. Epiligrin, the major human keratinocyte integrin ligand, is a target in both an acquired autoimmune and an inherited subepidermal blistering skin disease. J. Clin. Investig. 1992, 90, 1628–1633.
- Amber, K.T.; Bloom, R.; Hertl, M. A systematic review with pooled analysis of clinical presentation and immunodiagnostic testing in mucous membrane pemphigoid: Association of anti-laminin-332 IgG with oropharyngeal involvement and the usefulness of ELISA. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 72–77.
- Christiano, A.M.; Greenspan, D.S.; Lee, S.; Uitto, J. Cloning of human type VII collagen. Complete primary sequence of the alpha 1(VII) chain and identification of intragenic polymorphisms. J. Biol. Chem. 1994, 269, 20256–20262.
- Siprashvili, Z.; Nguyen, N.T.; Bezchinsky, M.Y.; Marinkovich, M.P.; Lane, A.T.; Khavari, P.A. Long-term type VII collagen restoration to human epidermolysis bullosa skin tissue. Hum. Gene Ther. 2010, 21, 1299–1310.
- Conget, P.; Rodriguez, F.; Kramer, S.; Allers, C.; Simon, V.; Palisson, F.; Gonzalez, S.; Yubero, M.J. Replenishment of type VII collagen and re-epithelialization of chronically ulcerated skin after intradermal administration of allogeneic mesenchymal stromal cells in two patients with recessive dystrophic epidermolysis bullosa. Cytotherapy 2010, 12, 429–431.
- Siprashvili, Z.; Nguyen, N.T.; Gorell, E.S.; Loutit, K.; Khuu, P.; Furukawa, L.K.; Lorenz, H.P.; Leung, T.H.; Keene, D.R.; Rieger, K.E.; et al. Safety and Wound Outcomes Following Genetically Corrected Autologous Epidermal Grafts in Patients With Recessive Dystrophic Epidermolysis Bullosa. JAMA 2016, 316, 1808–1817.
- Eichstadt, S.; Barriga, M.; Ponakala, A.; Teng, C.; Nguyen, N.T.; Siprashvili, Z.; Nazaroff, J.; Gorell, E.S.; Chiou, A.S.; Taylor, L.; et al. Phase 1/2a clinical trial of gene-corrected autologous cell therapy for recessive dystrophic epidermolysis bullosa. JCI Insight 2019, 4.
- Pendaries, V.; Gasc, G.; Titeux, M.; Leroux, C.; Vitezica, Z.G.; Mejía, J.E.; Décha, A.; Loiseau, P.; Bodemer, C.; Prost-Squarcioni, C.; et al. Immune reactivity to type VII collagen: Implications for gene therapy of recessive dystrophic epidermolysis bullosa. Gene Ther. 2010, 17, 930–937.
- Woodley, D.T.; Cogan, J.; Wang, X.; Hou, Y.; Haghighian, C.; Kudo, G.; Keene, D.R.; Chen, M. De novo anti-type VII collagen antibodies in patients with recessive dystrophic epidermolysis bullosa. J. Investig. Dermatol. 2014, 134, 1138–1140.
- Woodley, D.T.; Cogan, J.; Hou, Y.; Lyu, C.; Marinkovich, M.P.; Keene, D.; Chen, M. Gentamicin induces functional type VII collagen in recessive dystrophic epidermolysis bullosa patients. J. Clin. Investig. 2017, 127, 3028–3038.