Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Samir K Maji and Version 2 by Camila Xu.
The synucleinopathies exhibit differences in their clinical and pathological representations, reminiscent of prion disorders. Emerging evidence suggests that α-Syn self-assembles and polymerizes into conformationally diverse polymorphs in vitro and in vivo, similar to prions. These α-Syn polymorphs arising from the same precursor protein may exhibit strain-specific biochemical properties and the ability to induce distinct pathological phenotypes upon their inoculation in animal models. In this review, we discuss clinical and pathological variability in synucleinopathies and several aspects of α-Syn fibril polymorphism, including the existence of high-resolution molecular structures and brain-derived strains.
- α-synuclein
- amyloid
- polymorphs
- synucleinopathies
1. Misfolding and Aggregation of α-Syn
Monomeric α-Syn is an intrinsically disordered protein and tends to adopt multiple conformational states affected by solution conditions like pH, temperature, ionic strength, viscosity, etc. [1][9]. For instance, the presence of alcohols (ethanol) or fluoroalcohols (TFE or HFiP) induces the formation of β-sheet or α-helical partially folded structures of α-Syn, depending on the concentration and the type of alcohol used [1][9]. α-Syn was first isolated from the antisera raised against the cholinergic vesicle from Torpedo californica, an electric ray [2][159]. Due to its location at the nuclear envelope and presynaptic terminal, it was named synuclein [2][3][159,160]. α-Syn protein is encoded by the SNCA gene mapped to the human chromosome 4q21.3-q22 [3][160]. α-Syn was also discovered by Ueda et al. [4][161] during the study of amyloid plaques from the brains of patients with Alzheimer’s, in which they identified a non-amyloid-β component (NAC) in the plaques, which was derived from a precursor protein, NACP [4][161]. It was detected in all the tissues except the liver, and the highest concentration was found in the brain [4][161]. Later, it was found that NACP is a natively unstructured and human homolog of α-Syn [5][6][7][162,163,164]. Extensive biophysical and structural characterization revealed that α-Syn is a 140 amino acid protein with a molecular weight of ~14.4 kDa and pKa of 4.7 [8][165]. It is known to be involved in neurotransmitter release, vesicle trafficking, and SNARE complex assembly in the brain, though its exact physiological role is still obscure [3][8][160,165]. α-Syn consists of three domains, N-terminal, NAC, and C-terminal domains (Figure 1A). N-terminal of α-Syn (residues 1–60) is an amphipathic, lysine-rich, and lipid-binding domain, which interacts with the membranes [9][109]. It contains 11 aa repeats, including conserved KTKEGV hexameric motifs [9][109]. These repeats are conserved across species as well as among three synuclein members. Although α-Syn remains unordered in an aqueous solution, it adopts a helical structure involving N-terminus upon association with negatively charged small unilamellar vesicles or detergent micelles [9][10][11][109,166,167]. Interestingly, all the familial mutations of α-Syn also occur in the N-terminus region [12][13][14][15][16][17][18][19][83,84,85,86,87,88,89,90] (Figure 1B). The NAC domain of α-Syn (residues 61–95) forms the protein’s hydrophobic core and is prone to aggregation. This domain is responsible for the conversion of α-Syn from an unordered state to β-sheet-rich fibrils [20][21][168,169]. NAC is also part of the membrane-binding domain of the protein [11][167]. The conformational ensemble of α-Syn monomer indeed consists of structures that are similar to the membrane-bound state of α-Syn [22][170]. These contain partially folded helices involving N-terminuses and NAC domains similar to the 1XQ8 model [22][170], suggesting that such a type of folding might also be present in the early stages of aggregation. The C-terminal domain (residues 95–140) is flexible and predominantly consists of negatively charged amino acids [8][165]. The disordered carboxy-terminal part is also involved in the nuclear localization of α-Syn protein and its interaction with metal, small molecules, and proteins [23][24][25][26][27][171,172,173,174,175].
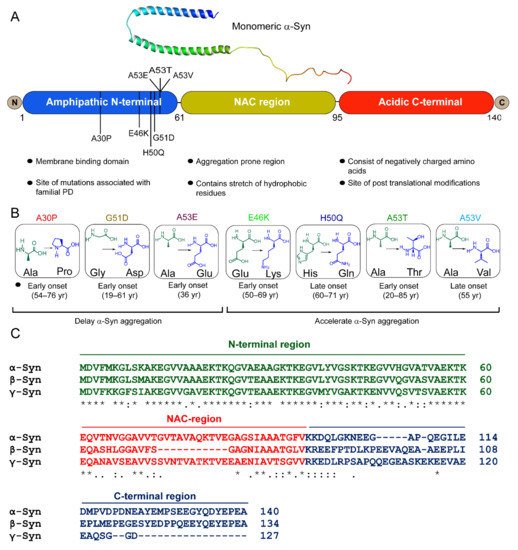
Figure 1. α-Syn structure and its disease-associated mutations. (A) Structure of micelle-bound human α-Syn [PDB ID: IXQ8] [11][167] and schematic representation of the primary sequence of α-Syn with three distinct domains, (i) the N-terminus (blue) contains lipid-binding motif and is the site of all familial mutations of α-Syn, and (ii) the central NAC domain (mustard) contains the stretch of hydrophobic residues. The two curved α-helices, helix-N (Val3-Val37) (blue) and helix-C (Lys45-Thr92) (green) in the micelle-bound α-Syn [1XQ8], connected by a short linker are formed within the 11 aa repeats (consensus sequence), which extends up to the first 89 residues [11][167]. (iii) the C-terminal (red) is rich in acidic amino acids. (B) Schematic diagram of seven mutational variants of α-Syn associated with familial PD along with their age of onset. A30P, G51D, and A53E delay and E46K, H50Q, A53T/V accelerate α-Syn amyloid formation. (C) Multiple sequence alignment of α-Syn, β-Syn, and γ-Syn by Clustal W. “*” indicates identical amino acids in all three variants, “:” and “.” indicate conserved and semi-conserved residues, respectively.
β-Synuclein (β-syn) and γ-synuclein (γ-Syn) proteins also belong to the synuclein family [8][165] (Figure 1C). β-Syn is 134 amino acid protein, earlier identified as the human homolog of bovine phosphoneuroprotein 14 (PNP14). The 11-amino-acid (residues 73–83) stretch is missing in its NAC domain (Figure 1C), due to which it lacks the ability to fibrillate [28][29][30][11,176,177]. Earlier, it was believed that β-Syn is an inhibitor of α-Syn aggregation and prevented its neurotoxicity [28][29][11,176]. However, this notion changed after discovering missense mutations in β-Syn gene, P123H (familial), and V70M (sporadic), known to cause DLB [31][178]. The deleterious effects of these mutations have been shown by cell- and animal-based studies [32][33][179,180]. Our group recently showed that under normal physiological conditions, fibrilization and aggregation of β-Syn and its disease-associated mutations did not occur, but an altered microenvironment, such as a decrease in pH and/or presence of heparin, caused them to polymerize [34][181]. γ-Syn, which shares ~55% sequence homology with α-Syn, was initially identified in breast cancer malignancies encoded by a breast-cancer-specific gene, BCSG1 [35][182]. It was reported in the peripheral central nervous system and breast cancer tissues [35][36][182,183]. It aggregates and forms fibrils in vitro [30][177] and in cells [37][184], but is comparatively slower than α-Syn [38][91].
The misfolding and fibrillation of α-Syn is a major event in several neurodegenerative disorders [39][185]. The misfolded α-Syn aggregates are amyloidogenic in nature, which aberrantly accumulate in the brain, and, as a result, the patient suffers movement abnormalities that worsen over time. The aggregation of α-Syn is a complex phenomenon and involves the conversion of monomers to highly ordered cross-β-sheet-rich structures through the formation of several soluble on- and off-pathway oligomeric species [39][40][185,186]. The amyloid formation of α-Syn is generally monitored by thioflavin T fluorescence dye [41][187]. It follows sigmoidal growth kinetics, which consist of (i) the lag phase, involving the formation of nuclei, which eventually grow into the detectable aggregate structure in solution; (ii) the elongation phase, the conversion and subsequent growth of oligomeric species into the fibrillar structure; and (iii) the stationary phase, representing the steady-state where the monomer and fibril concentration reaches the equilibrium. By the end of the aggregation, α-Syn assembles into atypical long amyloid fibrils, normally characterized by electron microscopy and atomic force microscopic imaging techniques [1][9]. These phases of aggregation cannot be attributed to a single event or microscopic process. Instead, all the processes, viz., primary nucleation, elongation, secondary nucleation, and fragmentation, are active through all the phases of the growth curve but at different rates [42][43][188,189] (Figure 2). These reaction rates are governed by aggregation rate constants and the concentration of the reacting species at a given time [44][190]. The amyloid formation initiates with primary nucleation of the monomeric species in the solution and elongation by addition of monomer to growing ends of the aggregates [43][189]. However, primary nucleation processes are short-lived and rapidly surpassed by secondary nucleation processes [45][191]. The fragmentation of the fibrils under agitation conditions (or even under quiescent conditions depending upon the stability of amyloid fibrils) modifies the number of growing ends and significantly affects the overall growth kinetics [42][43][188,189]. Moreover, secondary nucleation by surface catalysis is also one of the major contributors to amyloid growth in several systems, especially under quiescent conditions [43][44][189,190].
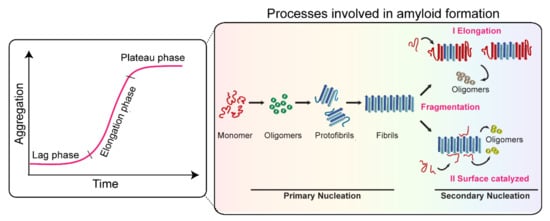
Figure 2. Processes involved in α-Syn aggregation. The amyloid growth kinetics of α-Syn follow three different phases, (i) the lag phase, (ii) the elongation phase, and (iii) the stationary/plateau phase. Primary nucleation, secondary nucleation, fragmentation, and elongation processes are active through all the phases of the growth curve, however, at different rates.
The self-assembly and aggregation of α-Syn is a complex phenomenon and involves multiple parallel processes. Therefore, it is crucial to understand the underlying molecular events to delineate their fundamental connection with human disease.
2. Prion-like Strain Phenomena in α-Syn
Since the discovery of α-Syn as the main constituent of Lewy body pathology in 1997, the primary focus has been shifted in delineating the underlying pathogenic mechanism of PD. Heiko Braak [46][192] presented a staging system of Lewy pathology in 2003 based on the specific patterns of α-Syn spreading. According to the Braak hypothesis, the Lewy pathology initiates from the olfactory bulb and DMV and then progressively spreads to the other brain regions. Although there is experimental and clinical evidence supporting Braak’s hypothesis, it is uncertain whether it is applicable and/or accurately describes the progression of PD in all the patients. For instance, there are cases in which patients do not show Lewy pathology in DMV or ENS, while other brain regions are severely affected [47][48][49][50][51][52][193,194,195,196,197,198]. Even in some cases, no link has been observed between the severity of Lewy pathology and clinical symptoms in PD [49][195]. Therefore, it is suggested to only apply Braak’s hypothesis to a subset of the population [52][198] as not all PD patients adhere to the staging system proposed by Braak [53][199]. Intriguingly, the reports of Lewy pathology in fetal neuronal grafts after fourteen years of transplantation into the striatum of the PD patient provided direct proof of cell to cell transmission and the spreading of α-Syn pathology proposed by Braak [54][55][200,201]. Studies using in vitro and cell model systems later suggested that α-Syn aggregates are infectious, can move from one cell to another, and seed the aggregation of their soluble endogenous counterpart in the recipient cells, explaining the phenomenon observed in grafted neurons [56][57][58][18,21,23]. This prion-like transmission of α-Syn aggregate from one region to another is also implicated in DLB and PDD patients, suggesting that the spread of Lewy pathology is the shared property of α-Syn aggregates in synucleinopathies [59][60][61][62][202,203,204,205]. However, the clinical and pathological features of these synucleinopathies are highly variable and heterogeneous [63][64][206,207], [65][66][146,208]. One might ask, why, despite being linked to the aggregation of the same protein, the distribution of α-Syn pathology and the manifestation of disease symptoms are different amongst synucleinopathies. This could be explained by the prion-like strain phenomenon of α-Syn, in which the same precursor protein forms different fibrils that result in distinct pathology.
2.1. Concept of Prion Strains
The last few decades of research have suggested that proteins/peptides with various structures and sequences can form a common fold of cross-β-sheet-rich structure of amyloid [67][68][69][70][71][209,210,211,212,213]. These proteins/peptides form amyloids with a common aggregation framework, i.e., through nucleation-dependent polymerization mechanism [72][73][214,215]. However, each protein/peptide may also undergo a distinct aggregation pathway to form a unique amyloid structure. Recent high-resolution structural studies with ssNMR and cryo-EM have indeed suggested that each protein packs uniquely and forms different structures for the cross-β-sheet fold [74][75][76][77][78][56,57,58,61,216]. Not only that, but, surprisingly, one protein can form multiple different structural folds [79][80][40,59]. Thus, these proteins can adopt various conformations from the same amino acid sequence, giving rise to several proteinopathies and, therefore, not confirming the one protein–one structure hypothesis [81][217]. For instance, tau folds differently in Alzheimer’s and Pick’s disease [79][82][83][40,218,219]. Different TAR DNA-binding protein (TDP-43) aggregates exist in the brains of Frontotemporal lobar degeneration (FTLD-TDP) subtypes, showing morphological differences across the subtypes [84][220]. This protein’s ability to misfold and display conformational diversity can lead to severe consequences, such as neurodegeneration [85][221]. This phenomenon of a protein to form different amyloids associated with various phenotypic properties is well known for prions [86][87][46,222]. Prions are infectious protein particles that show conformational heterogeneity and can be transmitted from one individual to another [88][223]. A myriad of evidence shows epidemiological and clinicopathological diversity in human prion diseases, such as Kuru disease, Gerstmann–Straussler–Scheinker syndrome, and Creutzfeldt–Jakob disease [89][224], as well as non-human prion diseases, such as bovine spongiform encephalopathy (BSE) in cattle, scrapie in sheep and goats, etc. [90][225]. The normal cellular prion protein (PrPC) undergoes conversion from α-helical to β-sheet-rich conformation (PrPSc), which is an insoluble, PK-resistant, and infectious form. PrPSc propagates and aggregates following two widely accepted mechanisms/models, i.e., the template-assisted and nucleation polymerization model. A pathogenic prion acts as a template in the template-assisted model and provides a surface for converting an endogenous normal prion protein to its misfolded pathogenic form [91][226]. In the nucleation–polymerization model, monomeric PrPSc combine and form a stable nucleus, also called a seed. These seeds keep on recruiting PrPC and convert them to their pathogenic counterparts [91][226]. One of the remarkable properties of prions is that they can misfold into diverse conformations, each giving rise to distinct clinical, histological, and pathological profiles. These aggregates with different conformations and pathological behavior are referred to as ‘strains’ [92][93][45,227]. The pioneer observations on the presence of prion strains came from the study by Pattison and Milson, 1961 [94][228], wherein they experimentally produced scrapie in goats and observed distinct clinical manifestations of the disease owing to different strains. In another study, Fraser and Dickinson were able to distinguish different strains of scrapie in infected mice models depending upon the extent of damage in different regions of the brain [95][229].
Despite resulting from the aggregation of the same prion protein, the prion diseases differ from each other with respect to the disease onset/incubation period, progression, and histopathological lesions in the infected brain [89][90][224,225]. This has been collectively termed as ‘prion strain phenomena’ [92][86][96][45,46,238]. Recent studies, however, have suggested that this strain property of amyloids is not only limited to prions but other amyloids associated with various neurodegenerative disorders such as Alzheimer’s and Parkinson’s [97][98][99][19,246,247], [56][100][18,248]. Various evidence has been provided from in vitro and in vivo studies to demonstrate the prion-like strain behavior of α-Syn, as discussed below.
2.2. α-Syn Strains Generated In Vitro
Growing evidence of prion-like strains of α-Syn associated with clinical and pathological variations observed in synucleinopathies has been reported in the past few years [101][102][103][104][105][106][107][108][29,30,31,32,33,34,35,249]. These strains have been defined as distinct and stable conformational assemblies of a single protein that can self-replicate and propagate in vivo and result in different disease phenotypes. Guo et al. discovered two ‘strains’ of α-Syn pre-formed fibrils (pffs), through de novo fibrilization, termed as ‘strain A’, and repetitive seeding fibrilization in vitro, termed as ‘strain B’. These two strains exhibited distinct conformations and striking differences in cross-seeding tau protein in primary neuronal cultures and in vivo [102][30]. Besides documenting the evidence of α-Syn fibril strains for the first time, these findings also identified the cross-seeding behavior of α-Syn. Later, several groups took advantage of the chameleon property of α-Syn [1][9] and screened numerous growth conditions to generate α-Syn strains in vitro. Under different growth conditions, several conformationally stable and unstable states of protein were observed [109][250]. The conformations, which are not thermodynamically stable or cannot establish stable intermolecular interactions, cannot grow into amyloid fibrils. These are referred to as growth-incompetent states (Figure 3). On the other hand, growth-competent states of α-Syn can grow and form different fibrils depending on the growth conditions (Figure 3). The resulting fibrils under different assembly conditions not only possess different biochemical and biophysical properties, like resistance to proteases, cytotoxicity, seeding ability, etc. [101][102][103][104][106][107][110][111][112][29,30,31,32,34,35,36,51,53], but also imprint their architecture on the daughter fibrils depending on the growth condition and the nature of seeds (Figure 3). Bousset et al. indeed generated two structurally and functionally different α-Syn strains, named ‘fibrils’ and ‘ribbons’, using different physiological salt concentrations [101][29]. Fibrils caused more cytotoxicity, whereas the ribbons were found to be more effective in inducing α-Syn inclusions in vivo [105][33]. Similar differences were obtained with strains generated by Suzuki et al. [111][51], where one strain caused the accumulation of abundant phosphorylated and ubiquitinated α-Syn aggregates in cultured neurons and mice due to its ability to interact with proteasome complexes, whereas the other strain failed to do so [111][51]. Nowadays, varying experimental conditions have rather become a common strategy to generate strains in vitro. In cases where the criteria to be called a ‘strain’, i.e., should be a structural variant of the protein aggregates, exhibit the ability to self-propagate and serially transmit the disease over the next generations and cause clinical and phenotypical disease variations, are not completely fulfilled, it would be more appropriate to call the fibrils as ‘polymorphs.’ These polymorphs may show striking differences in their morphology and structure [113][74][114][28,56,251], nucleation rates [115][252], seeding and membrane binding ability in cells [110][36], etc. However, as a functional consequence of these structural variations in polymorphs, they may or may not result in distinct clinical subtypes of diseases. There are also cases of intrasample polymorphism, which can arise irrespective of whether fibrils are generated in vitro [116][75][117][118][119][120][39,57,253,254,255,256] or derived from brain extracts [79][121][122][40,257,258]. While these may possess certain commonalities, like a similar monomeric fold or share a common structure, they also show marked differences in morphology, β-strand arrangement, or biochemical properties [75][120][57,256]. Thus, different assembly conditions can generate structurally and functionally distinct fibrillar assemblies, which may either propagate as a unique α-Syn strain or may simply form polymorphs.
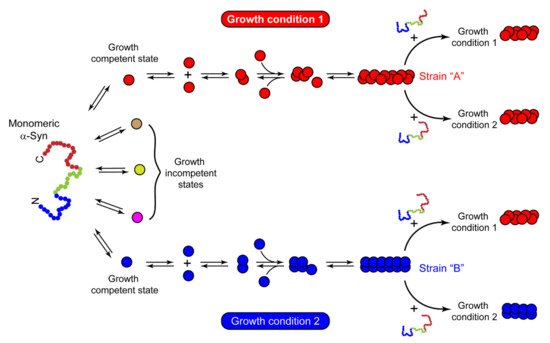
Figure 3. Mechanism of strain formation and its propagation. Natively unstructured monomeric α-Syn populates into multiple conformational states with distinct abilities to form amyloid fibrils. Red and blue circles represent the growth-competent states, which, under certain sets of growth conditions 1 and 2, form different fibril strains ‘A’ and ‘B’, respectively. Strain ‘A’ seeds the monomeric protein and passes on its structural architecture to the next generation of fibrils, irrespective of growth condition 1 or 2. In contrast, strain ‘B’ passes its structural characteristics on the daughter fibrils only in the same assembly conditions that were used for its growth (growth condition 2). This suggests that the nature of seeds and growth conditions play an essential role in deciding the fate of assemblies upon cross-seeding. The other conformations in brown, yellow, and pink circles cannot yield thermodynamically stable intermolecular interactions and are incapable of growth. These are referred to as growth-incompetent states.
2.3. α-Syn Strains in Human Synucleinopathy Samples
The structural and functional differences observed in recombinant strains can be validated by identifying and characterizing fibrils directly from synucleinopathy patient samples. The first evidence of a brain-derived strain came from a seminal study by Prusiner et al. [123][270], which demonstrated that brain extracts from MSA are transmissible to transgenic mice and cells, resulting in abundant α-Syn pathology [123][270]. In contrast, this was not observed using brain extracts from PD, suggesting that the PD-derived strain may differ from MSA [123][270]. Then comes the question, what might lead α-Syn to adopt a different conformation in MSA or PD? In vitro, various solution conditions (like the presence or absence of salt) give rise to fibrils with different structural and functional properties [101][29]. Similarly, α-Syn is also exposed to several microenvironments in vivo, affecting its aggregation [124][271]. The dopaminergic neurons in PD and the oligodendrocytes affected in MSA belong to different cell lineages and have distinct cellular environments. Lee and co-workers demonstrated that distinct intracellular environments of two cell lines impart strain formation in MSA and PD [106][34]. α-Syn fibrils derived from GCIs in oligodendrocytes (GCI-α-Syn) and LBs in neurons (LB-α-Syn) of diseased brains differ significantly and exhibit distinct seeding abilities [106][34]. GCI-α-Syn strain is highly effective in seeding α-Syn aggregation compared to LB-α-Syn, thereby contributing to the aggressiveness of MSA [106][34].
α-Syn aggregates have also been detected in biological fluids like cerebrospinal fluid (CSF) and plasma of PD patients [125][126][272,273]. α-Syn aggregation begins years before the onset of actual disease symptoms and, thus, the detection of these aggregates at early stages may enable the identification and characterization of a particular strain in these fluids. In this context, the amplification of α-Syn aggregates from brain extracts of PD and MSA patients using the protein misfolding cyclic amplification (PMCA) technique has been recently developed. This technique involves the amplification of misfolded proteins in vitro, in a manner similar to DNA amplification by PCR [127][274]. It consists of alternate cycles of incubation and sonication, resulting in amyloid replication. First, the trace amount of amyloid is incubated with an excess of native protein to induce polymer growth. Then, the sample mixture is subjected to sonication, which will break down the fibrils, resulting in several nuclei. Each newly formed nucleus will then act as a seed in the next cycle and further induce the growth of fibrils. This way, after each cycle, the number of seeds will increase exponentially and will allow the detection of the minute amount of misfolded aggregates present at the beginning [127][274]. Soto and co-workers used PMCA to amplify the α-Syn aggregates from the CSF of the patients diagnosed with PD and MSA [128][52]. They found that PD- and MSA-derived fibrils exhibit different biophysical and biochemical properties and correspond to distinct conformational strains of α-Syn [128][52]. Even α-Syn aggregates amplified from PD and MSA brain homogenates have been shown to exert variable toxicity and neurodegeneration in human dopaminergic neurons, reflecting different disease severity observed in PD and MSA patients due to different strains of α-Syn [129][275]. These findings conclusively suggest that synucleinopathies can be distinguished based on the type of α-Syn strain present in the brain. However, the complexity in detecting aggregates arises when patient-to-patient heterogeneity is observed in the same disease. This heterogeneity in α-Syn aggregates amplified from PD patients’ brain extracts is greater than MSA brain extracts [130][276]. Strohaker et al. reported that the fibrils derived from PD and MSA do not exhibit markedly distinct structural properties [130][276], in contrast to findings reported by Soto and co-workers [128][52]. The possible reason for the contrasting observations could be the differences in the PMCA protocols used by the two groups [131][277]. Additionally, Strohaker et al. used a much smaller sample size than Soto’s group [128][130][52,276]. Other factors, like the genetic background of the patients, age of the selected patients, a load of α-Syn aggregates in different patients, presence of other components in the extracts, and region of the brain from where extraction was done, could also be responsible for these differences [130][276]. Similar contradictions also exist in the field of AD pathology. Recent findings on brain-derived tau samples have suggested patient-to-patient heterogeneity in the tau fibril conformations exist within the same disease, AD [132][278]. However, Goedert and colleagues observed the same type of tau conformation in all AD cases analyzed so far, suggesting that tau fibrils from a single disease (like AD or Pick’s disease) adopt a common structural fold [82][83][218,219]. Although the reasons and factors that drive this structural specificity in tauopathies are unclear, it could be due to multiple isoforms of tau, PTMs, interactions with other protein molecules, co-factors, etc. Recently, Scheres and Godert presented a hierarchal classification of tau fibrils from different tauopathies based on the folds of their filaments [133][279]. Whether a similar classification exists for α-Syn fibrils isolated from synucleinopathy samples remains to be determined. Recently, a great effort has been made to solve the structure of α-Syn derived from the human brain by Schweighauser et al., using Cryo-EM [134][280]. The group found that α-Syn filaments from the brain of DLB patients do not twist and are thinner than those derived from the brain of MSA patients [134][280], consistent with the previous findings [135][3]. The lack of twists in fibrils derived from DLB precluded the determination of 3D structure by cryo-EM and the differences in α-Syn fibrils derived from MSA and DLB patients were drawn based on two-dimensional class averaging [134][280]. Although we need more high-resolution structures derived from synucleinopathy patients to reach a definite conclusion, the present reports certainly strengthen the claims on the existence of distinct fibril types of α-Syn. Moreover, the structures of α-Syn filaments from PD cases are not yet available, but solving them in the future can significantly help to understand the disease mechanism and generate therapeutic approaches against synucleinopathies.
3. Familial Mutations of α-Syn Form Distinct Fibril Conformations
α-Syn oligomerization and aggregation are associated with PD pathogenesis. Seven familial missense mutations have been discovered so far in the SNCA gene, associated with early- and late-onset PD [12][13][14][15][16][17][18][19][83,84,85,86,87,88,89,90]. Among these PD-associated mutations, E46K, H50Q, A53T, and newly discovered A53V mutants accelerate the rate of α-Syn aggregation, whereas A30P, G51D, and A53E mutations slow down the aggregation kinetics in vitro [38][136][137][91,93,96]. However, the link between the rate of aggregation (in vitro) and the age of the disease onset (in vivo) is not straightforward [138][103]. Although oligomers formed during the early stages of aggregation kinetics are potentially toxic [139][286], only A30P shows faster oligomerization and delayed conversion of oligomers into fibrils [140][102]. G51D, on the other hand, exhibits slow oligomerization and slow fibril formation [141][142][287,288], yet is associated with the early onset of disease. Due to this complexity in the behavior of familial mutants, it is challenging to set up a unifying mechanism by which they cause the disease.
Previous reports have suggested the α-Syn adopts a helical structure upon binding with membranes in vivo [143][144][289,290]. Any single amino acid change in the N-terminus domain of α-Syn may alter the membrane-binding ability and increase the cytosolic concentration of the protein by promoting faster aggregation [145][146][291,292]. The membrane-binding data of familial α-Syn mutants from our laboratory [147][92] and others [148][142][146][149][110,288,292,293] have shown that H50Q, A53T, and E46K mutants exhibit increased membrane binding, while A53E, G51D, and A30P mutants exhibit decreased membrane binding. This suggests that, similarly to aggregation, the membrane-binding capability does not correlate with increased disease propensities by familial α-Syn mutations. Therefore, there is a lack of correlation between the aggregation and membrane-binding ability with the actual disease pathogenesis caused by the familial mutants of α-Syn. This raises the question of how a point mutation in a natively unstructured protein shows drastic differences in the disease onset and progression. We believe it could be possible that different α-Syn mutants produce different types and amounts of oligomers and also may uniquely alter the seeding capacity of wild-type protein [138][103]. That is why mutants affect not only the overall aggregation rate of the protein, but also the microscopic steps involved in the amyloid formation, i.e., initiation and amplification of α-Syn through secondary nucleation process [150][294]. Intriguingly, Lazaro et al. found that, despite having identical oligomerization propensity in cultured cells, A30P, E46K, H50Q, G51D, and A53T exhibit distinct abilities to form inclusions [151][295]. A30P showed a decreased propensity to form inclusions in cells, whereas the E46K and G51D mutant displayed an opposite effect [151][295]. Again, the inclusion formation in cells [151][295] did not correlate with the aggregation propensity of mutants in vitro [152][38][147][136][140][12,91,92,93,102]. Thus, addressing these questions about how wild-type α-Syn and its mutants contribute to the early and late onset of PD becomes important to understand the differential pathogenesis of synucleinopathies.
Fibril formation is highly sensitive to changes in the local and/or global microenvironment of the protein. This suggests that a single amino acid change can result in polymorphism due to different site-specific conformational dynamics, as shown for the wild type and fibrils of E46K, A30P, and A53T [153][296]. In this context, Knowles and co-workers recently studied the systematic comparison of α-Syn and its disease-associated mutants using biophysical techniques [154][297]. PD mutants generate fibril polymorphs with distinct morphology and secondary structures compared to the wild-type protein [154][297].