Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Ferdinando Carlo Sasso and Version 2 by Catherine Yang.
Gliflozins are a novel class of anti-hyperglycemic agents used for the treatment of T2DM that selectively inhibit the SGLT2 in the kidneys, leading to an insulin-independent lowering of blood glucose levels through an enhanced daily urinary loss of up to 100 g of glucose (200–300 kcal). At the same time, they produce a natriuretic effect by inhibiting sodium reabsorption.
- endothelial dysfunction
- gliflozins
- cardiovascular diseases
- heart prevention
1. Introduction
Gliflozins are a novel class of anti-hyperglycemic agents used for the treatment of T2DM that selectively inhibit the SGLT2 in the kidneys, leading to an insulin-independent lowering of blood glucose levels through an enhanced daily urinary loss of up to 100 g of glucose (200–300 kcal) [1][17]. At the same time, they produce a natriuretic effect by inhibiting sodium reabsorption [2][3][18,19].
The design of gliflozins resulted from structural modification of phlorizin, a naturally occurring O-glucoside and non-selective SGLT1/SGLT2 inhibitor. In 2013, the FDA approved the first member of the gliflozin class, canagliflozin, for the treatment of T2DM. Other compounds currently approved include dapagliflozin, empagliflozin, and ipragliflozin. They are well-tolerated drugs that reduce glycosylated hemoglobin (HbA1c) by ~0.5%–0.8% with a low risk of hypoglycemia, decrease BP pressure levels, and induce a weight loss of approximately 2 kg. As predicted by the considerably lower expression of SGLT2 in tissues other than the kidney, the side effects of gliflozins are generally confined to urinary tract infections, although ketoacidosis has been reported and, in 2017, the FDA raised concerns for canagliflozin use over an association of leg and foot amputations [4][20].
Three large CVOTs have demonstrated undeniable CV beneficial effects [5][6][7][21,22,23], respectively for empagliflozin, canagliflozin, and dapagliflozin, that go beyond those expected from simple glycemic control, the optimization of has demonstrated effective CV protection [8][9][24,25]. The favorable impact on other conventional risk factors may be involved, such as blood pressure (BP) (3 to 6 mmHg reduction in systolic and 0 to 2 mmHg drop in diastolic BP as consequence of diuretic/natriuretic effect), renal dysfunction (reduced albuminuria by decreased intraglomerular pressure and glomerular hyperfiltration), and body weight (average loss of 1–3 kg) [10][11][26,27]. Of importance, the SGLT2-I effect on this last parameter involves visceral adiposity and is not merely a reflection of fluid loss but rather associated with a significant reduction of adipose tissue mass, as demonstrated by bioimpedance spectroscopy [12][28]. Changes in lipid profile with a decrease in fasting and postprandial triglyceride levels have been described after six months of empagliflozin treatment in T2DM patients with established CHD, whereas a reduction in cholesterol levels has been demonstrated only in some animal studies [13][14][15][16][29,30,31,32]. The decrease in extracellular fluid and plasma volume with synergistic reduction of both afterload and preload may contribute to cardiovascular benefits, especially in diabetic individuals with impaired function of the left ventricle (LV), CHD, or congestive HF [17][33]. The importance of the ability of gliflozins to modify various cardiovascular risk factors is indirectly confirmed by the brilliant results of a recent multicenter study, in which a multifactorial intervention was shown to reduce mortality and non-fatal major adverse cardiovascular events (MACE) in diabetic subjects with high cardiovascular risk [18][34]. Nevertheless, post-hoc analyses of trials have demonstrated that even when adjusting for BP, lipid status, and HbA1c over time, the reduction in CVD death and HF hospitalization is preserved, suggesting the involvement of other mechanisms. Accordingly, a recent analysis of EMPA-REG OUTCOME trial found that the cardioprotective effect of empagliflozin was independent of the achieved level of risk factor control, even if the risk for cardiovascular events was inversely correlated with the number of controlled risk factors at baseline [19][35]. On the other hand, the modest improvements of cardiovascular risk factors associated to gliflozin use is very unlike to have contributed significantly to the beneficial outcomes in a period of months or few years.
The pathophysiology underlying gliflozin CV actions remains not completely understood, despite the active ongoing research. A lot of contributing mechanisms, both systemic but also direct on vasculature and heart, have been speculated or are still under current investigation. Among these, gliflozins have been associated with the improvement of endothelial dysfunction (ED), a critical initiator of the macro- and micro-vascular complications in T2DM patients with high metabolic risk [20][36] (Figure 1).
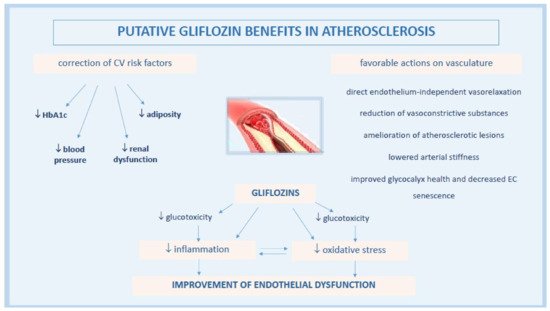
Figure 1. Pathophysiological mechanisms of anti-atherosclerotic actions by gliflozins.
2. Effects of Gliflozin on Vascular Endothelial Function
Impaired endothelial function plays a pivotal role in the pathophysiology of atherogenesis and is usually accompanied by increased oxidative stress and inflammatory responses [21][63]. In addition to atherosclerosis, ED has been associated with other types of CVD, including HF [22][23][24][25][64,65,66,67].
Accumulating experimental evidence in animal and in vitro models and related metanalyses [26][27][68,69] extensively document that gliflozin may modulate vascular EC activation and endothelial function, even if the expression of SGLT2 receptor on ECs and its role in the control of endothelial function remain interlocutory. Using an organ culture approach with murine aorta rings, immunohistochemistry and transport results showed that ECs per se are able to express the SGLT2 receptor [28][70]. A recent study further indicated that SGLT1 and SGLT2 protein levels are upregulated ex vivo in pathological arteries of adult rats (i.e., aortic arch exposed to disturbed flow and low shear stress and thoracic aorta exposed to Ang II or endothelial nitric oxide synthase (eNOS) inhibition), being responsible for ED most likely in consequence of the impaired endothelial formation of NO [29][71]. On the contrary, the inhibition of ROS generation and recovering of NO bioavailability induced by empagliflozin and dapagliflozin, as observed in human coronary arterial ECs (HCAECs) by Uthman et al., could not be attributed to the inhibition of SGLT2 as quantitative PCR to measure mRNA levels of this receptor resulted negative [30][72]. This observation is congruent with that of Mancini et al., who did not find SGLT2 mRNA in human umbilical vein endothelial cells (HUVECs) and HAECs [31][73].
Acute dapagliflozin treatment induced a direct vasorelaxation of abdominal aortic rings in ex vivo experiments from C57BI/6J mice maintained on a normal chow diet [32][74], a response not affected by the elimination of endothelium. Thus, an action on vascular smooth muscle cells cannot be excluded, as suggested by the vasodilator effect of dapagliflozin on rabbit thoracic aorta via the activation of protein kinase G (PKG) and thereby of voltage-dependent K+ (Kv) channels) [33][75]. In another study, empagliflozin was found to induce a vasodilation in thoracic aortic rings of rabbits similarly mediated by the activation of PKG and Kv channels, but not related to endothelial cells, other K+ channels, the cAMP/PKA pathway, Ca2+ channels, or sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) pumps [34][76].
The vasodilator effect of gliflozin could be vascular bed-dependent, as described for canagliflozin and the nonspecific SGLT-I phlorizin, which relaxed pulmonary arteries in a dose-dependent manner but had little or no effect on coronary arteries of C57BI/6J mice [35][77].
3. Glifozins and Heart Failure
Based on gliflozin positive findings from three main CVOTs, as well as from trials investigating CV benefits in chronic HF and two large multinational studies, SGLT2-Is have been identified as a new class of compounds for the treatment of diabetic patients suffering from HF, a common and serious comorbidity of T2DM whose prevention represent a crucial therapeutic goal [5][6][7][36][37][38][39][40][41][42][21,22,23,135,136,137,138,139,140,141].
Several possible explanations are suggested for gliflozin benefits in HF.
Merely, they correct factors predisposing or aggravating HF. Their pharmacological effects including osmotic diuresis induced by glycosuria and concomitant increased Na+ excretion, decreased BP and, more importantly, improved fluid overload with greater loss in interstitial relative to intravascular volume can reduce preload and LV filling pressures in HF patients [43][44][142,143]. Moreover, gliflozins may modulate the afterload through the reduction of arterial BP and stiffness [45][46][47][127,144,145].
The inhibition of SGLT2 may attenuate the downstream effects of neurohormonal activation in HF and ameliorate the tissue oxygenation thanks to an increase in hemoglobin and hematocrit levels [48][49][146,147].
Above all, gliflozins are being shown to exert numerous favorable actions on the heart, such as the amelioration of LV mass and hemodynamics, reduction of myocardial inflammation, oxidative stress and fibrosis, and direct pleiotropic effects on cardiomyocytes, although there is an apparent absence of detectable SGLT2 expression in the heart [50][51][52][16,148,149].
Based on recent evidence, a key point in the pathophysiology of EHpHF is the ED in microvascular coronary bed, another mechanism which may be beneficially affected by gliflozins (Figure 2).
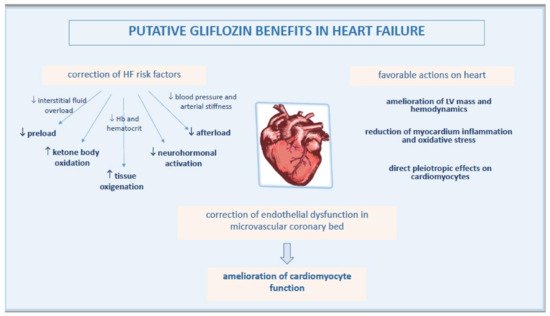
Figure 2. Pathophysiological mechanisms of improved heart function by gliflozins.
3.1. Gliflozins Correct Endothelial Dysfunction in Microvascular Coronary Bed
The impairment of endothelial integrity or function may represent a pivotal feature in HF, as indicated by high circulating levels of apoptotic endothelial-derived MPs, a surrogate biomarker and effector of ED, in non-diabetic patients with chronic HF [53][150]. Indeed, a clinical study showed that both ED and abnormal vascular structure, respectively indicated by FMD and intima-media thickness, were present in HFpEF and could contribute to its pathogenesis and maintenance [54][151].
Due to strict anatomical connection, a sick endothelium can affect cardiomyocyte health. Thus, the amelioration of endothelial function in coronary microcirculation may represent a key mechanism among those suggested in the protection of failing heart by gliflozins.
The mechanism by which SGLT2-Is could affect the function of cardiac microvascular endothelial cell is still debated [55][152]. It has been reported as an effect mediated by the inhibition of NHE1 in cardiomyocytes [56][57][153,154].
A study of the hearts of STZ diabetic mice revealed that empagliflozin improved myocardial structure and function by preserving cardiac microvascular barrier, sustaining eNOS phosphorylation and endothelium-dependent relaxation, as well as improving microvessel density and perfusion. These effects likely resulted from the inhibition of mitochondrial fission mediated by an AMPK-dependent mechanism inhibiting Drp1 activation [58][155].
In a rodent model of early diabetes, a 10-week treatment with empagliflozin improved coronary flow velocity reserve and fractional area change (a systolic contractility index) monitored with sequential measurement by non-invasive Doppler ultrasound imaging. The concomitant increase in l-arginine/ADMA ratio as an indicator of increased NO production, possibly indicated an improvement of cardiac contractile function via an NO-dependent improvement of endothelial coronary microvascular function [59][156].
Interesting results were obtained by Juni et al. using a novel co-culture system of human cardiac microvascular endothelial cells with adult rat ventricular cardiomyocytes. The study provided the first evidence that cardiac microvascular ECs modulated cardiomyocyte contraction and relaxation, an effect that was lost after pre-incubation with TNF-α but restored by empagliflozin through the reduction of ROS mitochondrial production and cytoplasmic accumulation, and by recovering endothelial NO bioavailability [60][81]. In a study on a similar model, the same investigators showed that uremic serum from patients on dialysis impaired endothelium-mediated enhancement of cardiomyocyte relaxation and contraction, an effect that was rescued by empagliflozin [61][157]. These results may provide a novel mechanism linking cardiac microvascular ED to the pathogenesis of HF in chronic kidney disease.
3.2. Gliflozin Benefit in HFpEF
The paradigm of HFpEF, a condition that accounts for a significant proportion of HF in the Western world, has shifted over the past years from a mere cardiomyocyte disease to a disorder that initially involves cardiac microvascular ECs. Currently, HFpEF is described as a systemic syndrome mediated by risk factors and co-morbidities, often of metabolic nature, resulting in a multi-organ pro-inflammatory state and leading to myocardial dysfunction and profibrotic remodeling through a cascade of events that initially involves cardiac microcirculation [62][158]. In this context, coronary microvascular dysfunction represents a prevalent phenotype and a powerful predictor of HFpEF, and ED a key determinant of its outcome [63][64][65][159,160,161].
An optimal level of intracellular NO might be critical for the proper function of cardiomyocytes, and nitrosative stress has been implicated as a master mediator in the pathophysiology of HFpEF [66][162]. Based on these premises and experimental findings, the improvement of myocardial microvascular EC function and amelioration of oxidative stress may represent the pivotal mechanisms by which SGLT2-Is prevent HF.
In a non-diabetic rat model of hypertension and diastolic dysfunction, which develops a series of metabolic, cardiac, and renal disturbances commonly observed in HFpEF patients, dapagliflozin mitigated some features of endothelial activation and dysfunction at the coronary level such as the upregulation of VCAM-1 and E-selectin, the downregulation of eNOS, and an increased expression of NFκB [67][163].
Two small investigations support these results in humans. In 184 diabetic subjects with HFpEF randomly divided into three groups (empagliflozin, luseogliflozin, and tofogliflozin), FMD was significantly higher and E/e’ (echocardiographic index of LV diastolic dysfunction) significantly lower at the end of a 12-week period of treatment, with a significant association between ∆mean E/e’ and ∆FMD [65][161]. In a pilot study evaluating 26 patients with T2DM and heart disease, a six-month treatment with tofogliflozin significantly ameliorated LV end-diastolic dimension but this change was not associated with a modification of %FMD, perhaps because this parameter was not suitable for the evaluation of small vessels, such as coronary microcirculation [68][164]. Incidentally, FMD was significantly improved only in patients with average HbA1c levels >8.0%, a result similar to the DEFENCE study reporting restoration of ED by dapagliflozin especially in diabetic patients with high HbA1c levels [69][121].