Monoamine oxidases (MAOs) are oxidative enzymes that catalyze the conversion of biogenic amines into their corresponding aldehydes and ketones through oxidative deamination. Owing to the crucial role of MAOs in maintaining functional levels of neurotransmitters, the implications of its distorted activity have been associated with numerous neurological diseases. Recently, an unanticipated role of MAOs in tumor progression and metastasis has been reported.
- monoamine oxidase
- cancer
- metastasis
- inhibitors
- QSAR
- pharmacophore
1. Introduction
By 1928, Mary Bernheim discovered the first enzyme of monoamine oxidase, and it was called tyramine oxidases [1]. Monoamine oxidases are in the flavin protein family, which is essentially composed of flavin amine oxidoreductases [2]. Monoamine oxidase can be classified into two types: monoamine oxidase A (MAO-A) and monoamine oxidase B (MAO-B). MAO-A is present in the gastrointestinal tract, lung, liver, and placenta, whereas MAO-B is present in blood platelets [3]. Monoamine oxidase A (MAO-A), a mitochondrial oxidative enzyme in the broad class of deaminating oxidases [4], essentially catalyzes the conversion of biogenic amines, such as dopamine and epinephrine, into their corresponding aldehydes through oxidative deamination with the concurrent production of reactive oxygen species (ROS) [5][6][7][5,6,7]. The prevalent existence of MAO-A is accredited to a conserved biological role in amine metabolism [5], and therefore it may be engaged in crucial cellular functions, such as monitoring cell growth and differentiation, maintaining the polyamine reservoir, and regulating levels of neurotransmitters [8].
Due to the essential role of MAO-A in preserving functional levels of neurotransmitters, the implications of its abnormal activity have been linked to several neurological disorders. For instance, increased activity of MAO-A has been associated with depression and anxiety [9][10][9,10]. On the other hand, deficiency of MAO-A enzymatic activity has been demonstrated in patients with mental retardation and abnormal behavior [11]. Moreover, the pathogenesis of neurodegenerative diseases, such as Parkinson’s and Alzheimer’s diseases, was proposed to be mediated by increased expression and activity of MAO-A [12][13][12,13]. Notably, several cardiovascular diseases, including heart failure [14], vascular remodeling [15] and myocardial injury [16], have been also related to abnormal levels and activity of MAO-A.
Monoamine oxidase B can be classified as a flavin adenine dinucleotide (FAD)-dependent mitochondrial enzyme. It acts primarily by catalyzing the oxidative deamination of different amines [17]. MAO-B shows preferential selectivity toward two substrates, which are 2-phenylethylamine and benzylamine. Dopamine and tyramine are also considered substrates for MAO-B. Selegiline (L-deprenyl), one of the first selective MAO-B inhibitors with an enhanced profile, has been used for almost 40 years to treat Parkinson’s disease. Glial brain cells are rich in MAO-B enzyme, especially in the neighborhood of dopaminergic synapses [18]. MAO-B participates mainly in storage regulation, release, and concentrations of biogenic amines in the synaptic cleft. Reactive oxygen species (ROS) are generated through monoamine substrate oxidation via MAO-B. Oxidation overabundance encourages neurotoxins synthesis. Tetrahyroisoquinolone (TIQ) and 6-hydroxydpamine are two of the most common neurotoxins whose high concentrations fasten chronic neurodegenerative disease [19].
During the last decade, several reports were published connecting the MAO-A-mediated production of ROS with tumor development and progression. Patients with advanced prostate cancer (PCa) showed elevated expression of MAO-A [20][21][20,21]. Oxidative stress and the resulting DNA damage caused by ROS were used to explain tumor initiation and progress in several types of cancers [22][23][22,23]. In addition, the ability of cancer cells to migrate from their primary location to another tissue appears to be linked to signaling pathways that involve MAO-A activity [24]. Interestingly, several MAO-A inhibitors were reported to modulate cell proliferation and result in cell cycle arrest in a dose-dependent trend. Cancer cell death induced by apoptosis pathways has been reported to be the main effect of certain MAO-A inhibitors on prostate cancer cells [25]. It is not only synthetic drugs that demonstrate anticancer activity; curcumin, a bioactive phytochemical compound that is proposed to work through the inhibition of MAO-A/mTOR/HIF-1α signaling pathways, revealed a reduction in cancer-associated fibroblast-induced invasion and ROS production in prostate cancer [26][27][28][26,27,28]. From this perspective, we review the existing studies on the role of MAO-A and MAO-B in cancer development and progression with a special focus on the design and use of MAO-A inhibitors in cancer chemotherapy.
2. Xanthone Derivatives
Gnerre et al. [29][96] studied MAO inhibitors in a set of 59 natural and synthetic xanthones derivatives ( Figure 13 ). The compounds showed more selectivity toward MAO-A than MAO-B with IC 50 values in the nanomolar range. Charge transfer interactions with the FAD cofactor are the most accepted hypothesis, although the molecular mechanism is not completely understood [30][97]. Both COMFA studies and ALMOND procedure are involved in studying the structural activity relationship [31][83].
Studying MAO-A activity by using topological descriptors, pyrrole derivatives are one of the examples of 2D-QSAR. La Regina et. al. studied a series of new pyrrole derivatives that are synthesized and evaluated for their monoamine oxidase (MAO) A and B inhibitory activity and selectivity [32][98].
It was found that the most selective compounds were N -Methyl, N -(benzyl), N -(pyrrol-2-ylmethyl)amine ( 5) and N -(2- benzyl), N -(1-methylpyrrol-2-ylmethyl)amine ( 6) ( Figure 24 ) for MAO-B [(1, SI) 0.0057] and MAO-A [(2, SI) 12500] inhibitors, respectively. Docking and molecular dynamics simulations play an important role in giving structural insights into the MAO-A and MAO-B selectivity. This could be explained by compound ( 6), as it forms a H-bond with Gln215 through its protonated amino group in the MAO-A binding site, while it is absent in the compound ( 5) MAO-A complex. Moreover, it could be noticed that 5 places its phenyl ring into an aromatic cage of the MAO-B receptor binding pocket, as it forms charge–transfer interactions. The slightly different binding pose of 6 into the MAO-B active site appears to be forced by a bulkier Tyr residue, which substitutes a smaller Ile residue present in MAO-A [33][99]. A study of 32 pyrrole derivatives ( 2) ( Figure 13 ) and analogues with 28 topological descriptors was accomplished using SPSS software, through multiple linear regression [32][98]. A model with a squared correlation coefficient (0.9) was found [32][98]. Seven topological descriptors were chosen through stepwise regression to be in the last model: the total structure connectivity index (Xt), mean square distance index (MSD), all-path Wiener index (WAP), eccentric index (DECC), Kier flexibility index (PHI), superpendentic index (SPI) and the mean Wiener index (WA) [34][100], as well as the cross-validation strategy, were investigated. It is found that the positive coefficients of the indices DECC, MSD, PHI, and SPI confirm that an increase in their values produces higher values for the Ki. However, large WA, WAP, and Xt (negative coefficients) decrease the Ki values [35][101].

For phenylalkylamine scaffold ( 4), the structural properties for 29 compounds were analyzed by Norinder et al. and a set of physiochemical descriptors were calculated ( Figure 13 ) [36][104]. Different partial least square (PLS) models were generated with squared correlation coefficients (r 2) with a value greater than 0.85. The authors concluded that the most essential SAR and high in vivo and in vitro activities require (S)-stereochemistry and no substitution on the aliphatic chain. In order to develop the best QSAR, electronic descriptors are essential variables. In order to increase in vivo activities, it is essential to attach small, electron-withdrawing and hydrophilic substituents in ortho and meta positions. While symmetrical, electron-withdrawing and lipophilic substituents in the ortho position are important for in vitro activity [36][104]. The order of NHMe > NMe 2 > NH 2 > CHMe 2 in para positions decreases both the in vivo and in vitro activity of the compounds [36][104].
3. MAO Inhibitors
MAO inhibitors differ in their origin; some are naturally available, while others are derived synthetically as shown in Table 1 . Interestingly, MAO inhibitors show a different pattern of selectivity toward MAO-A and MAO-B with variant IC 50, such as clorigyline with an IC 50 value of 0.0049 µM toward MAO-A.
| Scaffold | Selectivity | IC50 MAO-A | IC50 MAO-B | Chemical Structure |
|---|---|---|---|---|
| Chalcones [37] | Nonselective | 43.4 µM | 43.9 µM | 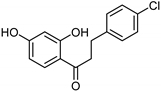 Xanthoangelol 13 |
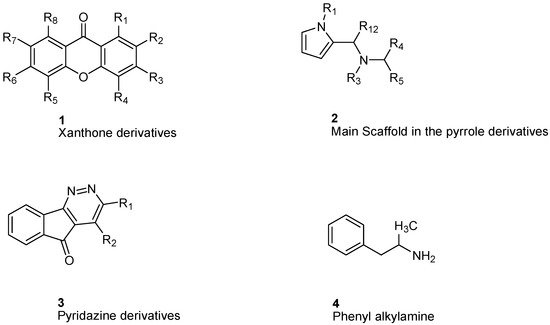
| Flavonoids [37] | Selective | 1.23 µM | NA | 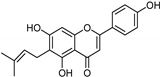 14 |
| Coumarins [37] | Nonselective | 8.9 nM | 8.9 nM |  15 |
| Xanthones [37] | Nonselective | 13.92 µM | 13.92 µM | 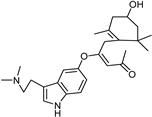 Desmodeleganine 16 |
| Nicotinamide [37] | Selective | 0.045 µM | 26 µM | 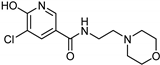 17 |
| Caffeine [37] | Selective | 34 µM | 0.148 µM | 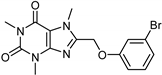 18 |
| Indole alkaloids [37] | Selective | 0.07 µM | NA | 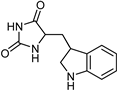 19 |
| Anthraquinone [37] | Selective | 2.5 µM | NA | 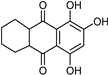 Purpurin 20 |
| Synthetic [38] | Selective | 5.5 nM | 150 nM | 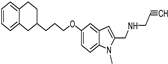 21 |
| Synthetic [29] | Selective | 0.49 µM | NA | 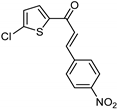 22 |
| Synthetic [29] | Selective | 0.14 mM | NA | 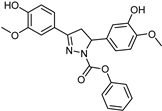 23 |
| Synthetic [29] | Selective | 0.06 µM | NA | 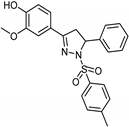 24 |
| Synthetic [29] | Selective | 0.01 µM | 2.15 | 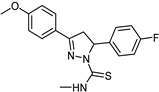 25 |
| Synthetic [29] | Selective | NA | 20 nm | 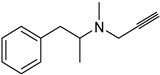 Selegiline 26 |
| Natural [29] | Selective | NA | 8.3 μM | 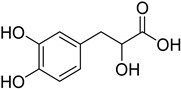 Danshensu 27 |
4. MAO-A Inhibitors as Anticancer Agents
MAO-A is a novel target gene of repressor element-1 silencing transcription factor (REST). It was reported that the neuroendocrine differentiation (NED) of prostate cancer (PCa) requires the downregulation of REST and activation of autophagy [39][126]. Studies showed that MAO-A inhibitors (pargyline and phenelzine) significantly reduced the NED and autophagy activation of PCa cells. Therefore, MAO-A inhibitors were considered a potential therapy for neuroendocrine tumors [39][126]. Yang et al. reported that a combination of isoniazid (INH), MAO-A inhibitor and tumor-targeting hepatomethine cyanine dyes proved to be a highly promising treatment tool for advanced PCa [40][127]. On the other hand, clorgyline, a selective irreversible inhibitor of MAO-A, prompted a mesenchymal to epithelial transition (EMT) in MDA-MB-231. Biological data showed that clorgyline induced E-Cardin (known epithelial protein marker) in breast cancer (MDA-MB-231) cells.
Moreover, clorgyline was shown to interfere with the β-catenin/p-GSK3β complex in addition to the E-cadherin/β-catenin complex. Overall, MAO-A is an essential regulator of EMT in breast cancer. Contrastingly, clorgyline was shown to reduce temozolomide (TMZ)-resistant glioma progression. Clorgyline induced cytotoxicity and reduced tumor cell invasion [41][39]. Thus, MAO-A inhibitor either alone or in combination with a low dose of TMZ may be potential therapy for the treatment of brain tumors [41][39].