The mammalian high temperature requirement A (HtrA) proteins are a family of evolutionarily conserved serine proteases, consisting of four homologs (HtrA1-4) that are involved in many cellular processes such as growth, unfolded protein stress response and programmed cell death. In humans, while HtrA1, 2 and 3 are widely expressed in multiple tissues with variable levels, HtrA4 expression is largely restricted to the placenta with the protein released into maternal circulation during pregnancy. This limited expression sets HtrA4 apart from the rest of the family. All four HtrAs are active proteases, and their specific cellular and physiological roles depend on tissue type. The dysregulation of HtrAs has been implicated in many human diseases such as cancer, arthritis, neurogenerative ailments and reproductive disorders.
1. Introduction
The high temperature requirement A (HtrA) proteins are a family of evolutionarily conserved serine proteases that are found in a large number of organisms ranging from prokaryotes, yeasts, and fungi to plants, birds, fish, and mammals
[1][2][1,2]. A unique feature of HtrAs is the presence of a trypsin-like protease domain in combination with one or more C-terminal domains called PDZ [postsynaptic density protein 95 kDa (PSD95),
Drosophila disc large tumour suppressor (Dlg1) and zonula occludens-1 (ZO-1)]
[2]. PDZ is a structural motif found in many signalling proteins and is known to participate in protein–protein interactions
[3]. The PDZ domain in prokaryotic HtrAs facilitates substrate binding and regulates oligomer assembly, cellular localization, and protease activities
[2]. According to the current MEROPS database, HtrA proteases belong to the C subfamily of the S1 family peptidases (chymotrypsin) within the PA clan, with the catalytic triad composed of histidine, aspartate and serine residues. The protein DegP (degradation of extracellular proteins) from
Escherichia coli is the first HtrA identified, which is characterised to be an ATP-independent serine protease with heat shock-induced proteolytic activities and a chaperone function
[4][5][4,5]. To date, over 180 HtrAs have been found across prokaryotic and eukaryotic organisms
[6]; in general, they play a key role in protein quality control by recognising the severely mis-folded/damaged proteins and targeting them for degradation, and many but not all also function as chaperones
[1]. The chaperone activity of HtrA/DegP involves multimer oligomerisation to form a cage-like structure, which facilitates the packaging and internalisation of mis-folded proteins to partition them between refolding vs degradation pathways
[7]. HtrA/DegP can switch between the two pathways to determine whether a misfolded/unfolded protein can be rescued or degraded, thus promoting protein stability and homeostasis
[8][9][8,9].
Prokaryotic HtrAs play an essential role in bacterial virulence and survival under cellular or environmental stress
[10][11][12][13][14][15][10,11,12,13,14,15]. Plant HtrAs maintain the photosynthetic machinery by acting as a chaperone in the assembly of photosystem II and as a protease in the degradation of photo-damaged reaction centres
[2]. In mammals, four HtrA homologs (HtrA1-4) have been identified across many species, and they participate in various cellular processes, such as cell growth, unfolded stress response, programmed cell death and aging
[1][2][16][1,2,16]. The dysregulation of HtrAs has been implicated in diverse pathological processes, such as cancer, neurogenerative disorders, arthritic disease and reproductive disorders
[17][18][19][20][21][22][23][24][17,18,19,20,21,22,23,24]. A recent review has summarised the current understanding of human HtrAs and their activation mechanisms from a structural perspective
[16]. Here, we discuss up-to-date knowledge of the four human HtrAs regarding their key molecular characteristics, tissue distribution, potential substrates and distinctive implications in various human diseases especially cancer and pregnancy complications. Animal studies will also be discussed whenever appropriate.
2. Protein Domain Architecture, Tissue Distribution and Key Molecular Characteristics of Human HtrAs
2.1. Domain Architecture
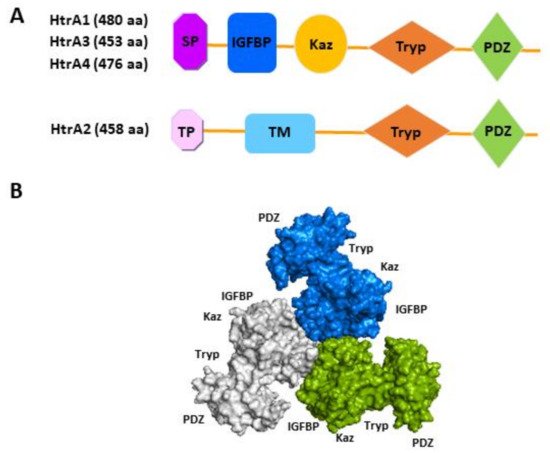
All four human HtrAs contain the signature motif of a HtrA, a chymotrypsin-like serine protease domain and a C-terminal PDZ domain (
Figure 1)
[25]. However, their N-terminal organisation differs considerably, naturally separating the family into two distinctive groups (
Figure 1A). While the N-terminal region of HtrA1, HtrA3 and HtrA4 contains a putative signal peptide, an insulin-like growth factor binding protein (IGFBP) domain and a kazal protease inhibitor domain
[25], that of HtrA2 is entirely different, encompassing a transient peptide and a transmembrane domain (
Figure 1A)
[26]. Deviations from the above basic domain organisation, resulting from alternative mRNA splicing or post-translational modifications, have been reported or inferred for HtrA1, 2 and 3
[16]. For instance, a shorter isoform of HtrA3 lacking the PDZ domain is expressed in the human endometrium and placenta, and a similar mRNA alternative splicing has been reported for HtrA3 in other species such as the mouse and rat
[26][27][28][26,27,28]. Alternative splicing will be discussed in the subsequent molecular characteristics section.
Figure 1. Schematic illustration of protein domain organisation of the four human HtrAs. (
A) The distinctive N-terminal region of HtrA2 compared to the other three HtrAs; (
B) The predicted trimeric form of HtrA3 (Adapted from Singh, et al., 2014)
[25]. SP, signal peptide; IGFBP, insulin growth factor binding protein domain; Kaz, Kazal-type S protease inhibitor domain; Tryp, trypsin-like serine protease domain; PDZ, postsynaptic density protein 95,
Drosophila disc large tumour suppressor and zonula occludens-1 domain; TP, transient peptide; TM, transmembrane domain; aa, amino acid.
The domain architecture of human HtrA2 resembles that of bacterial HtrA protein DegS
[16], whereas the N-terminal IGFBP-Kazal tandem module present in HtrA1, 3 and 4 is a new addition
[16]. It is noteworthy that the IGFBP-Kazal tandem is exclusively found in these HtrAs and another three mammalian proteins (Mac25, Kazal D1, IGFBPL1)
[29]. These differences suggest that HtrA1, 3 and 4 may have evolved to fulfil more mammalian specific cellular and physiological functions, whereas HtrA2 may have retained certain features of the ancient HtrAs. Indeed, studies suggest that although all four human HtrAs are active proteases and all form a trimeric or higher oligomeric assembly as prokaryotic HtrAs (
Figure 1B), the regulation of oligomerisation and protease activity of HtrA2 resembles that of prokaryotic HtrAs, whereas that of HtrA1, 3 and 4 is distinctly different
[16]. For instance, the PDZ domain of HtrA2 critically mediates its proteolytic activity, whereas HtrA1 and HtrA3 are active without the PDZ domain
[16]. It remains to be elucidated how the unique N-terminal IGFBP-Kazal tandem influences the physiological functions of HtrA1, 3 and 4. To date, a structural and functional analysis has found no evidence that these modules in HtrA1 retain their prototypic functions—the IGFBP region neither binds to IGFs nor behaves like an IGFBP, and the Kazal-like segment does not affect the HtrA1 proteolytic activity as would be expected of a protease inhibitor
[29].
2.2. Tissue Distribution
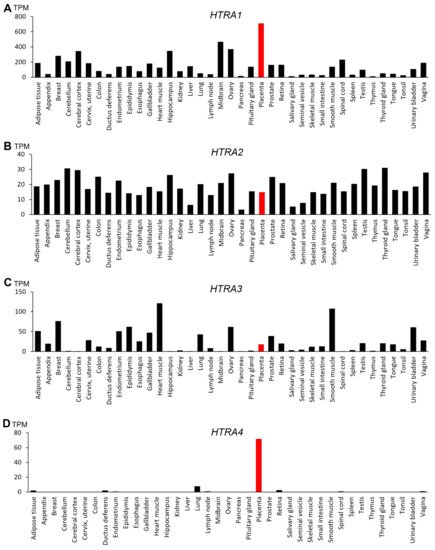
The four HtrAs have distinctive tissue expression in the human body (
Figure 2).
HTRA1 is widely expressed with the highest level detected in the placenta (
Figure 2A), suggesting that it may have an important role in placental development and function
[30][31][32][30,31,32]. The expression of
HTRA2, on the other hand, is ubiquitous and relatively uniform across many organs (
Figure 2B), though one study reported that the foetal liver expresses the highest level of
HTRA2 [26]. Like
HTRA1,
HTRA3 is also broadly expressed with high levels detected in the heart, smooth muscle, breast and the developing placenta (
Figure 2C)
[26][33][26,33].
Figure 2. Gene expression profile of the four human HtrAs in 39 human tissues. Data obtained from National Centre for Biotechnology Information FANTOM5 dataset and expressed in transcripts per kilobase million (TPM). The placenta is highlighted in red. (A) HTRA1; (B) HTRA2; (C) HTRA3; (D) HTRA4.
In contrast,
HTRA4 is not well expressed in humans, except in the placenta (
Figure 2D)
[24]. Moreover, placental
HTRA4 expression is likely human/primate-specific, as no other species are reported to express
HTRA4 abundantly in any tissues including the placenta. Liu et al. (2015) reported that
Htra4 null mice have normal embryonic and placental development, with no obvious differences in placental structure or morphology compared to the wild-type
[34]. Cross breeding of
Htra4 knock-out mice produces similar pup numbers, further indicating that fertility is unaffected
[34]. It is thus concluded that HtrA4 does not play a significant role in murine placentation or other HtrA family members have compensated for HtrA4
[34]. However, another explanation could be that
Htra4 is not well expressed in mice, including the placenta, which is somewhat supported by this study, although future research needs to confirm this possibility. Regardless, in the human
HTRA4 is highly expressed only in the placenta, setting HtrA4 apart from the other HtrAs. Studies to date suggest that HtrA4 exerts an important role in human placental development and pregnancy health, and in the pathogenesis of pregnancy complications
[24][35][36][37][38][39][40][24,35,36,37,38,39,40], which will be discussed in detail later in this review.
2.3. Key Molecular Characteristics
Table 1 lists the key molecular features of the four human HtrAs.
HTRA1 was initially identified as a gene downregulated in SV-40 transformed fibroblasts
[41]. Located on chromosome 10q26.13, the gene consists of 9 exons and encodes a polypeptide of 480 amino acids with a mass of approximately 50 kDa (
Table 1). Although predominantly considered as a secretory protein, HtrA1 with a mass of 29 kDa due to processing has been detected in the cytoplasm and within the nucleus of some tissues
[42]. The
HTRA2 gene is located on chromosome 2p13.1, containing 8 exons and encoding a protein of 458 amino acids and 49 kDa in size (
Table 1). HtrA3 was first identified as a pregnancy-related serine protease that is upregulated in the mouse uterus coinciding with placental development
[27]. In the human, the
HTRA3 gene is located on chromosome 4p16.1, comprising 10 exons which can be alternatively spliced to produce two protein isoforms—HtrA3L and HtrA3S
[26]. The long variant (
HTRA3L) lacks exon 7 and encodes for 453 amino acids (49 kDa), whereas the short variant (
HTRA3S) lacks exons 8, 9 and 10 and encodes for 357 amino acids (38 kDa, missing the PDZ domain)
[26]. HtrA4 was first identified as a serine protease associated with pregnancy
[1]; the gene is located on chromosome 8p11.22, encompassing 9 exons which encode a protein of 476 amino acids with approximately 50 kDa in size (
Table 1).
Table 1. Key features of human HtrAs.
Table 2,
Figure 3). For the signal peptide, HtrA1 shares 88% similarity with HtrA4, but only 50% with HtrA3, whereas HtrA3 and HtrA4 are not comparable (
Table 2). For the IGFBP domain, HtrA3 is 58% similar to HtrA1 or HtrA4, whereas the similarity is only 50% between HtrA1 and HtrA4 (
Table 2). Both the Kazal protease inhibitor domain and the PDZ domain are relatively more conserved, showing around 70% similarity among the members (
Table 2).
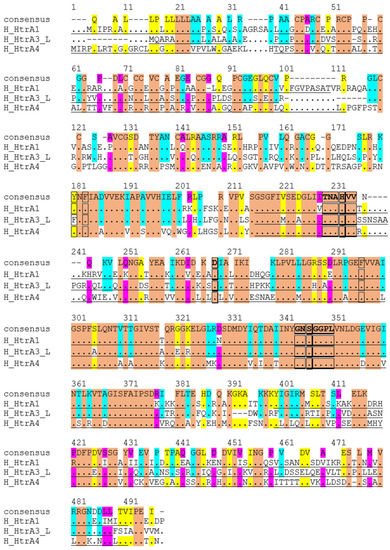
Figure 3. Alignment of amino acid sequences of human HtrA1, 3 and 4. Conserved amino acids are coloured: Orange, identical across all three HtrAs. Blue, similar between HtrA1 and HtrA3. Yellow, matching between HtrA1 and HtrA4. Pink, alike between HtrA3 and HtrA4.
Table 2. Comparison of amino acid sequences of human HtrA1, 3 and 4 proteins.
| Comparision between HtrA Members |
HtrA1 vs. HtrA3 |
HtrA1 vs. HtrA4 |
HtrA3 vs. HtrA4 |
| Identity |
Similarity |
Identity |
Similarity |
Identity |
Similarity |
| Approved name |
HtrA serine peptidase 1 |
HtrA serine
peptidase 2 |
HtrA serine peptidase 3 |
HtrA serine peptidase 4 |
| Overall |
58% |
73% |
54% |
| HtrA1 | 70% |
51% |
68% |
Degrading ECM proteins—fibronectin, type I collagen and decorin |
Musculoskeletal diseases |
[47] |
Gene name |
HTRA1 |
HTRA2 |
HTRA3 |
HTRA4 |
| Individual |
| domains |
Signal peptide |
50% |
50% |
89% |
89% |
| Processing ECM proteins—EFEMP1 and TSP1 |
Age-related macular degeneration |
[52][53] | [52,53 | − |
] | − |
Gene Synonyms |
ARMD7, CARASIL, CADASIL2, HtrA, IGFBP5-protease, L56, ORF480, PRSS11 |
MGCA8,
OMI,
PARK13,
PRSS25 |
TASP,
PRSP |
|
| IGFB |
52% |
58% |
45% |
| Processing ECM proteins—LTBP-1 |
Cerebral small vessel disease
CARASIL |
[46][54] | [46,54] | 50% |
Entrez Gene ID |
5654 |
27,429 |
94,031 |
203,100 |
| 50% |
58% |
| Kazal |
58% |
72% |
52% |
68% |
48% |
67% |
| Degrading APP and tau protein aggregates |
Alzheimer’s disease |
[43][57] | [43,57 |
HGNC ID |
9476 |
14,348 |
30,406 |
26,909 |
| ] |
Trypsin |
77% |
89% |
73% |
Degrading XIAP to activate caspase activity
Disruption of microtubules to inhibit cell migration |
Cancer |
[58][59] | [58,59] | 88% |
69% |
81% |
Ensembl ID |
ENSG00000166033 |
ENSG00000115317 |
ENSG00000170801 |
ENSG00000169495 |
| PDZ |
| Processing ECM proteins or growth factors involved in trophoblast migration and invasion | 41% |
Preeclampsia71% |
41% |
68% |
[31][37][60 | 41% |
] | 70% |
[31,37,60] |
MEROPS ID |
S01.277 |
S01.278 |
S01.284 |
S01.285 |
| HtrA2 |
Degrading unfolded or misfolded proteins |
Parkinson’s disease |
[61] |
Chromosoml location |
10q26.13 (122,461,553–122,514,907)
Plus strand |
2p13.1
(74,529,405–74,533,556)
Plus strand |
4p16.1
(8,269,712–8,307,098)
Plus strand |
8p11.22
(38,974,228–38,988,663)
Plus strand |
| Gene size (bases) |
53,355 |
4152 |
37,387 |
14,436 |
| [ | 62 | ] | [61,62] |
| Breaking down APP in mitochondria to maintain normal cellular function |
Alzheimer’s disease |
[63] |
| Binding to and degrading IAPs to facilitate caspase activities |
Cancer and chemoresistance |
[64][65] | [64,65] |
RefSeq |
NM_002775
NP_002766 |
NM_013247
NP_037379 |
NM_053044
NP_444272 |
NM_153692
NP_710159 |
| mRNA (bases) |
2091 |
1785 |
2539 |
2032 |
| Number of exons |
9 |
8 |
10 |
9 |
| Alternative splicing |
2 additional variants predicted |
9 additional varaints predicted with some confirmed |
1 additional variant confirmed |
unknown |
| UniProtKB/Swiss-Prot ID |
Q92743 |
O43464 |
P83110 |
P83105 |
| Full lenght Protein (aa) |
480 |
458 |
453 |
476 |
| Transient peptide |
N/A |
1–31 (31aa) |
N/A |
N/A |
| Signal peptide |
1–22 (22aa) |
N/A |
1–17 (17aa) |
1–31 (31aa) |
| Propeptide |
N/A |
32–133 (102) |
N/A |
N/A |
Mature peptide
Mol_wt (dalton) |
23–480 (458aa)
49,048 |
134–458 (325aa)
34,981 |
18–453 (436aa)
46,945 |
32–476 (445aa)
47,685 |
| Transmembrane |
N/A |
105–125 (21aa) |
N/A |
N/A |
| IGF-binding domain |
33–100 (68aa) |
N/A |
21–77 (57aa) |
36–99 (64aa) |
| Kazal-like domain |
98–157 (60aa) |
N/A |
64–128 (65aa) |
88–154 (67aa) |
| Serine protease domain |
204–364 (161aa) |
166–342 (177aa) |
175–340 (166aa) |
202–362 (161aa) |
| PDZ domain |
365–467 (103aa) |
364–445 (82aa) |
359–444 (86aa) |
383–474 (92aa) |
Catalytic triad Histidine,
Aspartic acid and Serine |
H220
D250
S328 |
H198
D228
S306 |
H191
D227
S305 |
H218
D248
S326 |
| Trimer stabilization sites |
Y169
F171
F278 |
F149 |
Likely
F140
F142
F255 |
Likely
Y167
F169
F276 |
| Protein binding sites |
382.385,
387,
444.445,
448.449 |
361.364,
366,
423.424,
427.428 |
356.359,
361,
418.419,
422.423 |
380.383,
385,
442–443,
446.447 |
| IAP-binding motif |
unknown |
134-137 |
unknown |
unknown |
Potential
phosphorylation sites |
S195, T237,
Y238, T365,
S367, S456 |
T51, T242, T54, T326, S96, S330, T157, S400, S212,T453 |
S214,
S334,
T363 |
S310,
Y314,
S424 |
Given HtrA1, 3 and 4 are more evolutionarily evolved and share a similar domain architecture, the amino acid sequences of these three members are aligned (
Figure 3) and compared (
Table 2). Globally, they share around 50% identity and 70% similarity in amino acids (
Table 2). When individual domains are compared, the serine protease domain shows the highest conservation, with >80% residues being similar (
Table 2). In particular, the catalytic triad residues histidine, aspartic acid and serine, and the HtrA sequence motifs GNSGGPL and TNAHV, are completely conserved (
Figure 3). However, significant divergences are apparent outside the protease domain (
3. Function, Regulation, and Potential Substrates of Human HtrAs and Their Involvement of Various Diseases
3.1. HtrA1
HtrA1 has been implicated in a diverse range of pathological conditions, including cancer, age-related macular degeneration (AMD) and preeclampsia
[17][23][43][44][45][46][47][17,23,43,44,45,46,47]. Each of the HtrA1 subdomains and their specific functions have been reported by different studies with contradictory findings. Truebestein et al. (2011) reported that substrate binding can directly mediate the remodelling of the active sites of HtrA1 to induce its proteolytic activity, while the PDZ domain is dispensable for substrate attachment or activation
[48]. In contrast, Eigenbrot et al. (2012) showed that HtrA1 can exist in an active state in the absence of a substrate, whereby the substrate simply binds to the active HtrA1, and an unknown independent mechanism switches on its proteolytic activity to regulate equilibrium
[29]. In general, the distinct sequence of the PDZ domain is thought to play a role in recognition and binding of specific substrates, as well as in the regulation of protease activities
[49]. Studies of collagen C-propeptide cleavage showed that a binding of this substrate to the PDZ domain of HtrA1 is necessary to activate the protease activity
[50]. In the same study, HtrA2 failed to significantly bind to collagen C-propeptide, or exhibit any increased protease activity, demonstrating the specific nature of the substrate recognition, binding and enzyme activity of different HtrAs
[50]. Other domains of HtrA1 may also further refine the substrate specificity; one study shows that the serine protease domain and the preceding small linker region on the HtrA1 can mediate the binding and inhibition of transforming growth factor (TGF)-β and bone morphogenetic protein (BMP) signalling, whereas the PDZ domain is not involved in the binding or protein—protein interaction
[51].
To date, many physiological substrates of HtrA1 have been identified, implicating that HtrA1 has a wide range of biological functions in different tissues (
Table 3). Consequently, aberrant HtrA1 activity has been linked to various human diseases, and many growth factors and matrix proteins have been identified as potential HtrA1 substrates (
Table 3). For instance, HtrA1 is upregulated in various musculoskeletal diseases, coinciding with the increased fragmentation of several extracellular matrix (ECM) proteins that are known targets of HtrA1, which include fibronectin, type II collagen and decorin
[47]. Increased
HTRA1 expression is also implicated in AMD, which is closely associated with the increased degradation of various ECM proteins
[52][53][52,53]. On the other hand, decreased level of HtrA1 is linked to dysregulation of TGF-β signalling in cerebral small vessel disease, which can lead to early-onset stroke and dementia
[54]. As a hereditary disease, cerebral autosomal recessive arteriopathy with sub-cortical infarcts and leukoencephalopathy (CARASIL) has been identified to contain mutations within the
HTRA1 gene, which results in the loss of HtrA1 protein or reduced protease activities
[46][55][46,55]. One of the proposed mechanisms of HtrA1 action is to process latent TGF-β binding protein 1 (LTBP-1), an ECM protein that is required by TGF-β signalling. This is supported by the observation that fibroblast cells isolated from CARASIL patients and from
Htra1 knockout mice all show reduced LTBP-1 processing and attenuated TGF-β activity
[54]. However, an opposite effect of HtrA1 has also been reported, where it inhibits TGF-β signalling during bone development via degrading TGF-β type II and III receptors and preventing the activation of downstream TGF-β signalling
[56]. Indeed, embryonic fibroblasts isolated from mice with the
Htra1 gene deletion show an increased expression of TGF-β-induced genes, and the bone mass in these mice is also increased due to the upregulation of TGF-β activity
[56]. These data suggest that HtrA1 may regulate specific proteins/pathways in a tissue-dependent manner.
HTRA1 is also highly expressed in the brain and has been implicated in Alzheimer’s disease. HtrA1 can directly degrade various fragments of amyloid precursor protein (APP), and inhibition of HtrA1 activity results in the accumulation of beta-amyloid in astrocytoma cell culture media; these data suggest a potential role of HtrA1 in protecting the brain against accumulation of amyloid deposits, which is a major neuropathological feature of Alzheimer’s disease
[43]. Furthermore, HtrA1 is demonstrated to degrade the aggregated and damaged tau, a protein that aggregates into intracellular neurofibrillary tangles in many neurological disorders such as Alzheimer’s disease
[57]. In neuronal PC12 cells, HtrA1 mRNA and activity are upregulated in response to tau, and in patient brain samples, the extent of tau aggregates inversely correlates to the level of
HTRA1 expression; these results suggest an important role for HtrA1 in regulating protein quality control for neurological functions
[57].
Table 3. Potential functions and substrates of human HtrAs in association with various diseases.
| HtrA Member |
Functions and Substrates |
Associated Diseases |
References |
| Degrading HAX-1 to promote cell death |
| Mitochondria-related dysfunction |
| [ |
| 66 |
| ] |
| Degrading Ped-Pea15 to promote cell death |
Environmental stressor-induced cellular dysfunction |
[67] |
| Degrading WTP1 to increase c-Myc and JunB to promote apoptosis |
Cancer |
[68][69] | [68,69] |
| HtrA3 |
Cleaving ECM proteins—decorin and biglycan |
Osteoarthritis and cancer |
[70] |
| Cleaving cytoskeleton proteins—actin, β-tubulin and vimentin |
Destabilization of cytoskeleton dynamics in cancer treatments |
[71] |
| Acting as a chaperone by interacting with TCP1α chaperonin |
Cancer and alteration of cancer cells |
[71] |
| Cleaving XIAP to promote drug-induced apoptosis |
Cancer and chemoresistance |
[72] |
| Binding to BMP4, TGF-β1, TGF-β2 and GDF5 to inhibit their functions |
Placental development and
Preeclampsia |
[22][70][73] | [22,70,73] |
| HtrA4 |
Degrading fibronectin to impede trophoblast invasion |
Preeclampsia |
[74] |
| Cleaving endothelial junction protein VE-Cadherin to increase permeability |
Preeclampsia |
[40] |
| Cleaving VEGF-A receptor KDR to inhibit angiogenesis |
Endothelial dysfunction in preeclampsia |
[39] |
3.2. HtrA2
While HtrA1, 3 and 4 are all secreted proteins, the precursor of HtrA2 resides in the mitochondrial intermembrane space and serves as protein quality control to maintain mitochondrial homeostasis under normal physiological conditions
[75]. The loss of HtrA2 in mice leads to accumulations of unfolded proteins in the mitochondria, defective mitochondrial respiration, increased concentrations of reactive oxygen species (ROS) and neuronal cell death
[61]. Mice without the HtrA2 proteolytic activity due to a mutation in the
Htra2 gene, exhibit muscle wasting and neurodegeneration similar to symptoms of patients suffering from Parkinson’s disease
[62]. Several point mutations within the PDZ domain of the
HTRA2 gene have been identified in patients with Parkinson’s disease, and these mutations are demonstrated to result in HtrA2 inactivation and mitochondrial dysfunction in vitro
[76]. However, a large-scale population-worldwide genetic association study has failed to find a strong link between
HTRA2 variants and Parkinson’s disease, thus the relevance of HtrA2 activity in Parkinson’s disease remains to be further investigated
[77].
Under stressful conditions, HtrA2 can switch from a pro-survival factor to a proapoptotic player to facilitate cell death
[17]. Following stress stimuli, HtrA2 is released from the mitochondria into the cytosol, and subsequently binds to and degrades the inhibitor of apoptosis proteins (IAPs), thereby freeing up active caspases to induce apoptosis in the damaged or infected cells
[64][65][64,65]. Environmental stresses such as tunicamycin or heat shock all increase HtrA2 expression in mammalian cells
[78]. Cisplatin, a chemotherapeutic drug commonly used to treat various solid tumours, has also been shown to upregulate HtrA2 in a dose-dependent manner in both mouse and human renal cells; the knockdown of
HTRA2 by siRNA or a HtrA2-specific inhibitor renders renal cells resistant to cisplatin-induced apoptosis
[79]. Furthermore, HtrA2 also directly promotes cell death independent of the caspase pathway by degrading other antiapoptotic proteins, such as HS1-associated protein X (HAX)-1, proliferation and apoptosis adaptor protein 15 (PED-PEA15) and Wilms tumour protein 1 (WTP1)
[66][67][68][66,67,68]. Therefore, HtrA2 functions as a crucial mediator of cell survival as well as cell death. However, unlike the other three human HtrA members, HtrA2 does not appear to be involved in placental development or pregnancy complications. Protein substrates and human diseases that are associated with HtrA2 are summarised in
Table 3.
3.3. HtrA3
Like HtrA1, HtrA3 is secreted due to the presence of an N-terminal signal peptide
[26]. However, HtrA3 has also been detected in the mitochondria, and a processed form lacking the N-terminal domain has also been found in the cytoplasm, suggesting that the N-terminal region is necessary for HtrA3 transportation into the mitochondrion
[80]. A detailed analysis of the HtrA3 subdomains has identified that neither the N-terminal nor the PDZ domain is required for enzyme activity or substrate binding
[81]. However, the PDZ domain appears to be necessary for HtrA3 to form trimers; when the PDZ domain is removed both HtrA3S and HtrA3L are present as monomer, whereas HtrA3 with truncated N-terminal forms a stable trimer
[81]. This is unique to HtrA3, since both HtrA1 and HtrA2 remain trimeric following the removal of both the N-terminal and the PDZ domains, which in turn suggests that the trimer formation of HtrA3 is less stable and requires the PDZ domain to interact with other regions to maintain stability
[81]. Furthermore, in HtrA3S the C-terminal PDZ domain is replaced by a sequence of seven amino acids, yet its enzyme activity is not significantly impacted; it remains unknown whether this unique sequence has any specific roles
[81].
Both HtrA3 isoforms are proteolytically active and can function either as proteases or chaperones
[71][82][71,82]. Both isoforms can interact with cytoskeletal proteins such as actin, β-tubulin, vimentin and TCP1 chaperonin, which are important for actin and tubulin folding
[71]. While both HtrA3 isoforms can cleave these proteins and function as chaperones in vitro, HtrA3S has more efficient proteolytic activities, whereas HtrA3L is the most efficient HtrA protein in facilitating tubulin polymerization
[71]. Thus, the two HtrA3 isoforms may have different roles and function either as proteases or chaperones depending on the tissue type
[71].
Studies in mice have demonstrated that HtrA3 can bind to and inhibit various TGF-β superfamily members, including BMP-4, TGF-β1, TGF-β2 and growth and differentiation factor (GDF)-5
[70]. Both HtrA1 and HtrA3 can degrade ECM proteins such as decorin and biglycan, which are mediators of TGF-β signalling, suggesting that the two HtrAs may have complementary roles in the remodelling of ECM in specific tissues
[70].
Furthermore, HtrA3 functions as a proapoptotic protein in mitochondria-mediated cell death
[80]. Treatment of lung cancer cells with chemotherapeutic drugs etoposide and cisplatin causes autoproteolysis of HtrA3, leading to a product of 35kDa which lacks the N-terminal domain but contains the PDZ domain plus the full active protease domain, which is subsequently translocated from the mitochondria to the cytosol
[80]. This translocation of HtrA3 coincides with an increase in cell death, which can be attenuated by either suppressing
HTRA3 or overexpressing the anti-apoptotic factor B-cell lymphoma (BCL)-2
[80]. Moreover, forced
HTRA3 expression significantly reduces lung cancer cell survival following treatment with etoposide and cisplatin, whereas an inactive mutant form of HtrA3 has no impact, suggesting that the protease activity of HtrA3 is essential in modulating drug-induced cytotoxicity in cancer cells
[80]. However, the exact role of HtrA3 and its target substrates in programmed cell death are not well characterized. It is reported that both HtrA3 isoforms can bind to and cleave the X-linked inhibitor of apoptosis protein (XIAP) to significantly reduce its cellular levels in lung cancer cells when treated with etoposide, indicating a possible mechanism of HtrA3 action in promoting cancer cell death following chemotherapy
[72]. Both isoforms of HtrA3 without the N-terminal region are still able to cleave XIAP like their wildtype counterparts, suggesting that the N-terminal domain is not required for HtrA3 proteolytic activity
[72]. The potential substrates of HtrA3 and their roles in various human diseases are presented in
Table 3.
3.4. HtrA4
Examinations of various human tissues and cell lines have thus far detected abundant
HTRA4 expression only in the human placenta (
Figure 2) and BeWo cells (a choriocarcinoma trophoblast cell line)
[24]. Though the full physiological role of HtrA4 in the placenta remains to be investigated, studies to date suggest that HtrA4 promotes trophoblast invasion during placental development
[74][83][74,83]. In BeWo cells, when the endogenous
HTRA4 expression is knocked down, cell invasiveness is greatly reduced
[74]. In trophoblast-derived JAR cell line, the forced expression of the wild type
HTRA4 but not an protease-inactive mutant increases invasion
[74]. HtrA4 is shown to cleave the ECM protein fibronectin in vitro, indicating that it may facilitate cell invasion by disrupting the interaction between fibronectin and its integrin receptors that would otherwise impede trophoblast invasion
[74][84][74,84]. Glial Cells Missing-1 (GCM1), a placenta-specific transcription factor, is shown to upregulate HtrA4 to promote invasion, which can be inhibited by the transcriptional factor GATA3
[85]. GATA3 alone has no effect on HtrA4 expression, but when co-expressed with GCM1, it suppresses GCM1-mediated luciferase activity in 293T cells transiently transfected with a
HTRA4 reporter construct
[85]. However, this is an artificial system since 293T cells are not trophoblasts and do not endogenously express HtrA4. The knockdown of
GATA3 in BeWo cells elevates HtrA4 expression and increases cell invasive activity
[85]. In trophoblast cell line JEG-3, which expresses very low levels of
HTRA4, the knockdown of
GATA3 leads to a marginal increase in HtrA4, suggesting other unknown factors are required to induce HtrA4 expression
[24][85][24,85]. However, all above studies have used cell lines and it remains to be investigated if HtrA4 has a similar role in primary trophoblast cells and in vivo. HtrA4 was recently found to play an important role in trophoblast syncytialization, where its upregulation in primary trophoblast cells coincides with a surge in human chorionic gonadotrophin (HCG) release, which is a known indicator of syncytialization
[86]. Furthermore, silencing of the
HTRA4 gene in BeWo cells inhibits forskolin-induced syncytialization and HCG expression/secretion
[86].
Since HtrA4 is secreted out of the placenta and detected in the blood circulation of pregnant women, it could impact endothelial cells. Indeed, recombinant human HtrA4 can cleave the main surface receptor for vascular endothelial growth factor (VEGF)-A which is also known as kinase insert domain receptor (KDR), causing an inhibition of VEGF-A action and endothelial dysfunction
[39]. In addition, HtrA4 can cleave the main endothelial junctional protein vascular endothelial (VE)-cadherin, disrupting cell—cell connections and inducing intercellular gaps between endothelial cells
[40]. Furthermore, HtrA4 may cleave other cell surface receptors such as TGF-β type III receptor (TGFβRIII), producing soluble receptors that can inhibit TGF-β function
[87]. The involvement of HtrA4 in pregnancy complications such as preeclampsia will be further discussed in a later part of this review.
Studies have explored the potential role of HtrA4 in cancer cell lines, reporting that HtrA4 promotes cell death by degrading XIAP and pro-caspase 7, which was shown previously for HtrA1 and 3 in cancer cells
[88][89][88,89]. Several other proteins have also been identified as potential substrates of HtrA4, notably cytoskeleton proteins actin and tubulin, TCP1 chaperonin and S100A6 calcium-binding protein, through which HtrA4 may exert an effect on cytoskeleton homeostasis
[90]. These studies present potential insights into the possible mechanisms of HtrA4 action
[89][90][89,90]. However, so far, HtrA4 expression has not been shown in any primary cancer cells or in vivo tumours. The potential substrates of HtrA4 and its role in human diseases are summarised in
Table 3.