Dendritic cells (DCs) dictate the outcomes of tissue-specific immune responses. DCs instruct T cells to respond to antigens (Ags), including self-Ags, leading to organ damage in the context of autoimmune diseases, or to becoming regulatory T cells (Tregs) promoting and perpetuating immune tolerance. DCs can acquire tolerogenic properties in vitro and in vivo in response to several stimuli, a feature that opens the possibility to generate or to target DCs to restore tolerance in autoimmune settings.
- dendritic cells
- autoimmune diseases
- tolerance
- Autoimmune Diseases: Breakdown of Tolerance
1. Autoimmune Diseases: Breakdown of Tolerance
Breakdown of immunological tolerance can lead to unwanted and detrimental activation of immune responses against self Ags that causes autoimmune diseases, such as Type 1 Diabetes (T1D), Rheumatoid Arthritis (RA), Multiple Sclerosis (MS), and Inflammatory Bowel Disease (IBD) [1]. These pathologies, usually involving genetic predisposition and poorly defined environmental factors, are widespread. It is estimated that worldwide almost 1 in 10 individuals (7.6–9.4%) suffer from autoimmune diseases [2].
Organ destruction in autoimmune diseases is associated with the dysregulation of the immune system with hyper-reactive effector cells that escape the immune control mediated by the tolerogenic arm of the immune system, leading to hyperactivation of adaptive immune responses and chronic inflammation. Studies, in autoimmune patients, demonstrated that aberrant activation of immune cells, including lymphoid and myeloid cells, leads to inflammation in the target organs of autoimmunity [3][4][5][6]. Currently, approved therapies for autoimmune diseases involve lifelong administration of immunomodulatory and immunosuppressant drugs that can efficiently improve the outcomes of the disease but, on the other side, are often associated with severe side effects. Specifically, the drugs used in these treatments are non-specific and non-curative, and can induce a systemic, generalized, and persistent immunosuppression leading to the risk of chronic infections or cancer development [7]. The increased knowledge on the unique ability of DCs, able to induce primary immune responses bridging innate and adaptive immunity, revealed their central role in maintaining tissue homeostasis and tolerance. Further improvements in understanding how DCs promote immunological tolerance and the development of protocols for the manipulation of DC activity in vitro and in vivo will lead to actively explore the possibility of identifying effective and specific approaches based on DCs to cure autoimmune diseases. In this context, the use of DCs rendered tolerogenic by different means represents an attractive therapeutic approach for restoring permanent Ag-specific tolerance. Tolerogenic (tol)DCs can be used to specifically target the detrimental immune response against disease associated Ags, while they maintain the retention of the capacity of the immune system to be functional and reactive against other pathogens and malignancies [8]. In this review, we briefly introduce the different subsets of human DCs, and we review the involvement of DCs in the induction of tissue-specific autoimmunity. Furthermore, we present current approaches using tolDC-based therapies or targeting DCs in vivo for the treatment of tissue-specific autoimmunity.
- Dendritic Cells Are Central Players in Promoting Immune Responses in Autoimmunity
2. Dendritic Cells Are Central Players in Promoting Immune Responses in Autoimmunity
DCs in the immature state (iDCs) predominantly reside in the peripheral tissues and in secondary lymphoid organs [9][10][11] and serve as sentinels of the immune system, continuously patrolling the extracellular milieu. iDCs can recognize a plethora of pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) through the innate pattern-recognition receptors (PRRs), Toll-like receptors (TLRs) or c-type-lectin receptors. iDCs express high levels of PPRs and low levels of major histocompatibility complex (MHC) class II, CD80 and CD86, and their lysosomal activity is attenuated (reviewed in [12]). During classical immune responses, iDCs process the encountered Ags into smaller peptides, which can be presented on the cell surface in the context of MHC class I/II [13][14]. The encountering of the Ag drives the maturation of iDCs that lose their ability to process new peptides and they acquire the capacity to present Ags to T cells. Specifically, DCs upregulate the expression of MHC class II and co-stimulatory molecules (e.g., CD40, CD80 and CD86), secrete pro-inflammatory cytokines (e.g., IL-1β, IL-12, IL-6, and tumor-necrosis factor α (TNFα)) [15][16] and upregulate the expression of CCR7 and CXCR4, which enable them to migrate to lymph nodes, where they can present Ags to naïve T cells, driving their polarization toward pro-inflammatory Th1, Th2 or Th17 cells or CTLs (reviewed in [17]). For effective activation of T cells, three signals are required: (i) interaction between TCR and Ag/MHC complex; (ii) engagement of CD28 with co-stimulatory molecules (CD80 or CD86); and (iii) secretion of cytokines and chemokines.
Apart from the induction of efficient immune responses against invading pathogens and foreign Ags, DCs are critical modulators of both central and peripheral tolerance. During T cell development in the thymus, DCs play a critical role in the depletion of autoreactive T cells. DCs localized in the medulla, together with thymic epithelial cells, present self-Ags to thymocytes, and when the TCR/MHC interaction is strong, they promote self-reactive T cell apoptosis [18]. However, this mechanism does not fully assure the selection of T cell unresponsive to self and innocuous foreign Ags since: (i) self-reactive lymphocytes can escape negative selection; (ii) many innocuous environmental Ags, including those deriving from commensal microbiota, are not expressed in the thymus; and (iii) TCRs specific for foreign Ags can recognize MHC-self-Ag complexes. To overcome these events, in the periphery there are tolDCs which, by exploiting several immunosuppressive mechanisms, modulate the activity of potentially pathogenic T cells and promote the expansion or/and the differentiation of several subtypes of regulatory T cells, including classical CD4 + CD25 hi Foxp3 + Tregs [19][20][21] and CD49b + LAG-3 + type 1 T regulatory (Tr1) cells [22][23].
Four main mechanisms of peripheral tolerance have been described: induction of clonal anergy, metabolic modulation, secretion of anti-inflammatory cytokines and clonal deletion. TolDCs express low co-stimulatory molecules and high inhibitory receptors such as programmed cell death ligand (PDL)-1 [24] and inhibitory Ig-like transcripts (ILTs) [25][26]. These characteristics lead to T cell clonal anergy and T cell unresponsiveness due to Ag presentation in the presence of low co-stimulation, [27], or by the engagement of inhibitory receptors with their ligands expressed on the T cells. The latter include: PDL-1/PDL-2 interaction with programmed death 1 (PD-1) [28][29], the interaction between ILTs and classical and non-classical HLA class I molecules [30][31] and CD80/CD86 binding to the cytotoxic T-lymphocyte-associated protein 4 (CTLA-4). CTLA-4/CD80-CD86 interaction mediates CD80 and CD86 trans-endocytosis and degradation [32] or the induction of indoleamine 2,3-dioxygenase (IDO) [33][34], an enzyme that catabolizes tryptophan, an essential amino acid for T cell proliferation. IDO upregulation in tolDCs leads to: (i) T cell starvation by physical depletion of tryptophan from the local environment [35]; (ii) production of immune-toxic kynurenines that promote T cell apoptosis [36]; and (iii) accumulation of kynurenine, which, upon interaction with the aryl hydrocarbon receptor (AhR) on CD4 + T cells, promotes their polarization into Tregs [37]. TolDCs can also alter T cell responses by modulating the metabolic milieu through the expression of heme oxygenase-1 (HO-1), which catabolizes hemoglobin and promotes the production of carbon monoxide, overall reducing DC immunogenicity [38]. Moerover, tolDCs, by secreting anti-inflammatory cytokines (i.e., IL-10, TGF-β, and IL-35), are involved in promoting Treg differentiation. TolDC-derived IL-10 suppresses effector T cell responses and induces Tr1 cells [39]. IL-35, which can be secreted by DCs [40], promotes the differentiation of IL-35-producing FOXP3 + Tregs and suppress Th17 cell induction [41][42]. Also TGF-β can promote the induction of FOXP3 + Tregs [15]. In a preclinical model of MS, retinoic acid, a metabolite of vitamin A, has been used to modulate DCs that acquire the ability to induce Tregs and to inhibit Th17 cell polarization [43]. Finally, tolDCs by the expression of FasL and TNF-Related Apoptosis-Inducing Ligand (TRAIL) promote T cell clonal deletion [44][45].
- Strategies to Generate Ex Vivo Tolerogenic Dendritic Cells
3. Strategies to Generate Ex Vivo Tolerogenic Dendritic Cells
Several naturally occurring subtypes of TolDC have been identified in humans and the tolerogenic properties of DCs prompt researchers to set-up different protocols to generate ex vivo tolDCs for the treatment of autoimmune diseases (Figure 2Figure 1).
To keep DCs at immature state, Machen and colleagues designed a protocol based on the use of specific antisense oligodeoxyribonucleotide (AS-ODN) targeting CD40, CD80, and CD86 transcripts to suppress these proteins expression in murine bone-marrow derived DCs [46]. A single injection of AS-ODN-treated DCs into prediabetic NOD mice significantly delay the incidence of T1D without affecting the response of T cells to alloAgs [47]. These pre-clinical results lead to the translation of AS-ODNs to human monocyte-derived DCs and their clinical application in T1D patients. Administration of AS-ODN-treated DCs was well-tolerated with no observable adverse events or toxicities. In vivo treatment slightly increased the prevalence of FOXP3+ Tregs [48].
Dexamethasone (Dexa), a potent synthetic steroid, has been used to modulate the phenotype of DCs toward a tolerogenic state. Exposure of human CD14 + cells, during monocyte-derived DC differentiation, or murine bone-marrow precursors, to Dexa prevents the differentiation of fully maturated DCs, as evidenced by the altered expression of MHC, CD86, CD80 and CD40 molecules, and by the reduced IL-12 production and T cell stimulatory capacity [49]. RA patients derived Dexa-DCs were administered in knee joints and resulted in a reduced synovitis formation at 3 months after treatment [50]. In patients of MS, injection of autologous Dexa-DCs loaded with disease relevant Ags demonstrated to be safe and well tolerated. Finally, Dexa was also used in another clinical trial in combination Vitamin A for the treatment of Crohn’s Disease. In CD patients Dexa-VitA TolDCs were administered intraperitoneally and treatment was well-tolerated, and a clinical improvement was observed in one-third of the patients based on a CD activity index [51].
The active form of Vitamin-D3 (VitD3), 1,25-dihydroxyvitamin D3 (1,25(OH)2D3), impairs the differentiation of murine and human DCs in vitro and in vivo, leading to a tolerogenic phenotype characterized by low Ag presentation capacity, downregulation of costimulatory molecules, and inhibition of IL-12 production. VitD3-DCs are unable to fully activate T cells and to initiate an immune response [52]. In a clinical trial for T1D patients the intradermal injection of proinsulin-epitope loaded VitD3-tolDCs coincided with low grade toxicity not likely related to the therapy, with no signs of systemic immune suppression, no induction of allergy to insulin, no interference with insulin therapy, and no accelerated loss of β-cell function in patients with the remaining C-peptide[53] . VitD3-tolDCs loaded with a pool of 7 myelin peptides are used in two coordinated phase I clinical trials in MS patients, to test the safety and tolerability of autologous tolDCs-VitD3 and to compare two routes of tolDC administration, intradermal vs intranodal injection [54]. In an additional clinical trial, VitD3 and Dexa have been used in combination to generate TolDCs from RA patients’ monocytes. The cellular product was pulsed with autologous synovial fluid collected from inflamed joints and injected into knee joints of RA patients leading to an improvement of the clinical symptoms without worsening knee flares, or other side effects [50].
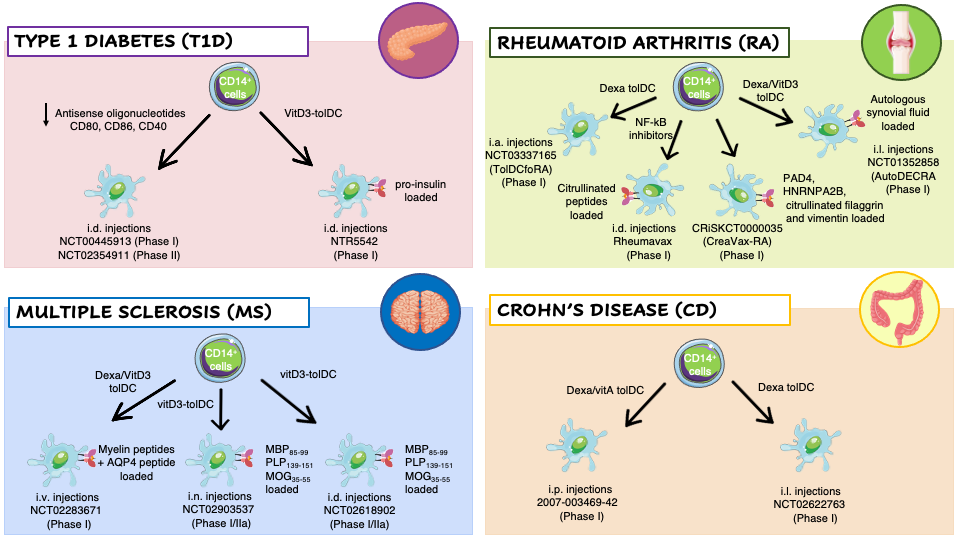
Figure 21. Clinical trials employing ex vivo generated tolerogenic DCs (tolDCs). TolDCs were differentiated starting from patients’ CD14+ monocytes in the presence of different tolerogenic agents. The resulting cells have been tested in clinical trials for different autoimmune diseases: Type 1 Diabetes (T1D) (pink panel), Rheumatoid Arthritis (RA) (green panel), Multiple Sclerosis (MS) (blue panel), and Crohn’s Disease (CD) (orange panel). Abbreviations: Dexa, Dexametasone; VitD3, 1,25-dihydroxyvitamin D3; VitA, vitaminA; NF-kB, nuclear factor kappa-light-chain-enhancer of activated B cells; AQP4, acquaporin-4; MBP85–99, Myelin Basic Protein; PLP139–151, proteolipid protein; MOG35–55, myelin oligodendrocyte glycoprotein; PAD4, peptidyilarginine deaminase 4; HNRNPA2B, heterogeneous nuclear ribonucleoprotein A2/B1; i.v., intravenous; i.n., intranodal; i.d., intradermal; i.p., intraperitoneal; i.l., intralesional; i.a., intra-articular.
- In Vivo Dendritic Cell Targeting to Mediate Tolerogenic Responses
4. In Vivo Dendritic Cell Targeting to Mediate Tolerogenic Responses
The ex vivo generation of TolDCs has some drawbacks including the extensive manipulation and the high manufacturing costs that prompted the researchers to exploit new approaches based on in vivo Ag-delivery to naturally occurring tolDCs or Ag co-delivered with immunomodulatory factors to polarize DCs toward a tolerogenic phenotype (Figure 12).
In the last decades, evidence demonstrated that Nanoparticles (NPs) were widely used in medicine to deliver drugs or molecules to specific cell subsets [55]. In the context of autoimmune diseases, DCs represent an attractive target for nanomedicine to directly promote their tolerogenic activity in an Ag-specific manner. Indeed, the combined delivery of tolerogenic agents and Ags into NPs promote DCs with the ability to present Ag in a tolerogenic manner [56]. To allow direct phagocytosis from DCs, NPs should have specific characteristics in terms of size, shape, and chemical properties [57]. In pre-diabetic NOD mice, injection of NPs encapsulating InsB and different tolerogenic agents (e.g., Vit D3 MPs + TGF-β1 MPs + GM-CSF) prevented T1D onset in 40% of the treated mice. Ex vivo analysis revealed decreased levels of insulitis in mice treated with tolerogenic NPs compared to controls, and an increased percentage of FoxP3 + Tregs [58]. NPs encapsulating antisense oligonucleotides targeting CD40, CD80 and CD86 molecules injected in NOD pre-diabetic mice knocked down the expression of these molecules. Interestingly, treatment with NPs encapsulating antisense nucleotides with or without InsB 9-23 in new onset NOD mice reverted diabetes [59]. Finally, intravenous administration of NPs encapsulated with p31Ag, a mimetope recognized by autoreactive T cells in T1D [60], preserved a normal islet architecture and prevented disease development induced by diabetogenic cells in NOD.SCID mice[61].
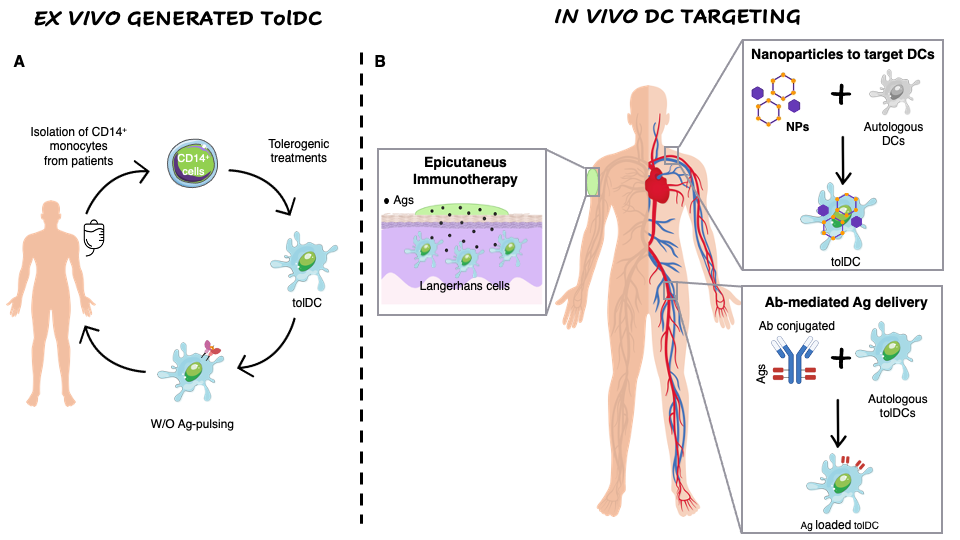
Figure 12. Ex vivo generation of TolDCs vs. in vivo DC targeting. (A) Ex vivo generation of TolDCs starts from CD14+ monocyte isolation from peripheral blood of autoimmune disease patients. Monocytes are differentiated into DCs in the presence of tolerogenic agents (i.e., Dexa, VitD3, AS-ODN CD80, CD40, CD86) and then pulsed or not with disease-relevant Ags. The obtained cellular product is infused in patients. (B) In vivo targeting of DCs. Nanoparticles encapsulating Ags w/o tolerogenic agents target autologous DCs and skew their phenotype toward a tolerogenic one. Antibodies fused to relevant Ags are generated to recognize surface markers expressed specifically by naturally occurring tolDCs (i.e., DEC205, DCIR2, CLEC9A). Epicutaneous immunotherapy that delivers the Ag to the APCs localized in the superficial layers of the skin.
The use of NPs to target DCs have been exploited also in preclinical models of MS, experimental autoimmune encephalomyelitis (EAE). Maldonado et al. demonstrated that injection of NPs encapsulating PLP, and RAPA inhibited EAE relapse [62]. Moreover, NPs encapsulating IL-10 and MOG injected subcutaneously in mice prior to the induction of EAE showed significant inhibition of EAE development together with a decreased frequency of CD3+ infiltration in the spinal cord of the treated mice compared to controls [63].
Freitag and colleagues demonstrated, in a mouse model of Celiac Disease (CeD), that intravenous infusion of gliadin-encapsulating NPs inhibited the proliferation, IFN-g and IL-17 secretion from gliadin-specific T cells, increased frequency of FoxP3+ Tregs, and decreased anti-gliadin antibody production [64]. Based on these promising results, two clinical trials have been performed to induce gluten-specific tolerance in CeD patients. Results showed that administration of gliadin-encapsulating NPs (TAK-101) was well tolerated, and that Ag-specific T cell response was reduced compared to placebo group. Furthermore, TAK-101 treatment was associated with a reduction in intestinal mucosa damages [65].
An alternative approach to induce Ag-specific tolerance is the use of Ag-delivering antibodies [66]. This system aims to selectively deliver a particular Ag to DCs using the highly specific binding of monoclonal antibodies (mAbs) to cell surface molecules expressed by naturally occurring tolDCs. Once injected in vivo, mAbs conjugated to an Ag, bind to their cognate ligand, are internalized and the delivered Ag is processed and presented by DCs to T cells. Since the purpose is to specifically target DCs with tolerogenic properties, the processed Ag will be presented in a pro-tolerogenic context, leading to the induction/expansion of Tregs, and/or anergy/deletion of Ag-specific effector T cells. Three types of Ag-delivering mAbs have been developed: chemical conjugates between native Abs and Ags; recombinant chimeric Abs; and single-chain fragment variable (scFv) constructs [67].
Due to the pro-tolerogenic properties of DEC-205 + BTLA hi DCs [68], the first recombinant chimeric Abs were originally designed to target DCs expressing DEC205 (CD205, LY75) [69]. Pioneer studies on this approach led to the establishment of efficient tolerance in different pre-clinical models of autoimmunity. In EAE, treatment with anti-DEC-205 chimeric Abs fused with disease relevant Ags resulted in amelioration of the disease score, prevention of pathogenic T cell accumulation in the CNS, induction of anergy in T effector cells, and reduction in IL-17 secretion[70][71]. Several studies in NOD mice demonstrated that anti-DEC-205 chimeric Ab or anti-DCIR2 chimeric Ab fused with insulin or β-cell-derived Ags induced clonal deletion of CD4 + and CD8 + autoreactive T cells, and conversion of pathogenic CD4 + T cells into FoxP3 + Tregs (reviewed in [67]). Furthermore, administration of anti-DEC205 coupled with a disease-relevant peptide reduced inflammation and symptom severity in models of proteoglycan-induced arthritis and Inflammatory Bowel Disease. At the cellular level, effector T cell deletion/anergy was induced and a portion of the autoreactive CD4 + T cells was converted into FoxP3 + Tregs [72][73]. An alternative target for Ag delivery to DCs is CLEC9A (DNGR1). Ag-coupled anti-DNGR-1 mAb promoted the proliferation of Ag-specific CD4 + T cells and their differentiation into Foxp3 + Tregs [74].
Epicutaneous immunotherapy (EPIT) is a novel immune-therapeutic approach that deliver the Ag to the APCs localized in the superficial layers of the skin via repetitive applications of an adhesive dermal patch containing a small amount of Ag (Figure 1). In animal models of food allergy it has been demonstrated that this approach induces desensitization to the given Ag, protects from inflammation and anaphylaxis, and induces Tregs (reviewed in [75]). Moreover, it has been demonstrated that the uptake of the Ag by Langerhans cells plays a central role in the induction of Ag-specific tolerance during EPIT [76]. Several clinical trials of EPIT have recently been completed or are ongoing for pollen, peanuts and milk allergies with encouraging results in terms of safety and tolerability (reviewed in [75]).
The work on the food allergies paved the way for the application of this approach also in autoimmune disease setting. In particular, promising results in preclinical models of MS [77] led to the development of two in-human study for EPIT in Relapsing Remitting MS patients that were treated with an adhesive patch containing a mixture of immunodominant myelin peptides applied to the skin. In the first study, transdermal immunization + promoted Tr1 cells and strongly suppressed myelin-reactive T-cell responses [78]. While in the second study, it was observed that the treatment was well tolerated and significantly reduced both magnetic resonance imaging outcomes (number of Gadolinium+ lesions) and clinical symptoms (relapse rate) [79]. The ability of EPIT to induce tolerance has been also tested in animal model of trinitrobenzene sulfonic acid (TNBS) induced ulcerative colitis. In the study the patches containing TNP-conjugated mouse immunoglobulin (TNP-Ig) were applied before the induction of colitis. Results showed that EPIT induced an amelioration of disease signs accompanied by a reduced production of IFNg and IL-17 and an increased production of IL-10 from solenocytes [80]. Finally, EPIT protocol has been also applied to Collagen Induced Arthritis (CIA) model. In this case the patches were soaked with type II collagen (COLL II) before CIA induction and their epicutaneous application was able to reduces disease severity [81].
- Overall Conclusions
5. Overall Conclusions
The improved knowledge of tolDCs and the development of protocols to generate cells ex vivo leads to the clinical application of these cells in autoimmune diseases. Overall, the generation of tolDCs ex vivo from patients’ monocytes is feasible and tolDC treatment can be safe and effective for some pathologies. However, there are some limitations that must be considered for improving the development of effective tolDC therapy. In particular, the selection of immune-relevant Ags is crucial, sometimes challenging or not applicable to all autoimmune diseases due to the lack of associated Ags. To overcome this problem, it has been proposed to pulse T cells with pools of different disease associated Ags [82]. Another important aspect to take into consideration is the specific migration of tolDCs to the disease target organ or to the relevant draining lymph nodes to enhance the therapeutic effect. To this end, different routes of administration have been exploited [83], or alternatively, the manipulation of tolDCs by over-expressing specific chemokine receptors to improve tissue-specific homing. Moreover, the generation of autologous ex vivo tolDCs requires the isolation and differentiation of the cells from patients’ monocytes, which may bear some alterations that will interfere with the functionality of the final cell product [84]. Finally, the production of ex vivo tolDCs requires extreme manipulation, which leads inevitably to high manufacturing costs. As an alternative to ex vivo manipulation, the development of alternative strategies to induce tolerance by autologous tolDCs in vivo can be considered.
Gaining knowledge on the biology of monocytes, the starting population used to generate ex vivo tolDCs, as well as on DCs in autoimmune diseases and on tolDCs, will lead to optimize the manufacturing protocols and to identify new approaches for the generation of innovative tolDCs and possible targets for in vivo modulation of DCs in the context of autoimmunity.
References
- Christopher Goodnow; Jonathon Sprent; Barbara Fazekas De St Groth; Carola Vinuesa; Cellular and genetic mechanisms of self tolerance and autoimmunity. Nature 2005, 435, 590-597, 10.1038/nature03724.
- Glinda S. Cooper; Milele L.K. Bynum; Emily Somers; Recent insights in the epidemiology of autoimmune diseases: Improved prevalence estimates and understanding of clustering of diseases. Journal of Autoimmunity 2009, 33, 197-207, 10.1016/j.jaut.2009.09.008.
- Frederique M Moret; Cornelis E Hack; Kim Mg Van Der Wurff-Jacobs; Wilco De Jager; Timothy Rdj Radstake; Floris Pjg Lafeber; Joel Ag Van Roon; Intra-articular CD1c-expressing myeloid dendritic cells from rheumatoid arthritis patients express a unique set of T cell-attracting chemokines and spontaneously induce Th1, Th17 and Th2 cell activity. Arthritis Research & Therapy 2013, 15, R155-R155, 10.1186/ar4338.
- Estifanos Ghebremedhin; Kelly Del Tredici; Mario Vuksic; Udo Rüb; Dietmar R. Thal; Guido J. Burbach; Albert Rosenberger; Heike Bickeböller; Thomas Deller; Rob A. I. De Vos; et al.Ernst N. H. Jansen SteurHeiko Braak Relationship of Apolipoprotein E and Age at Onset to Parkinson Disease Neuropathology. Journal of Neuropathology & Experimental Neurology 2006, 65, 116-123, 10.1097/01.jnen.0000199572.96472.1c.
- A Hänninen; S Jalkanen; M Salmi; S Toikkanen; Georgios Nikolakaros; O Simell; Macrophages, T cell receptor usage, and endothelial cell activation in the pancreas at the onset of insulin-dependent diabetes mellitus.. Journal of Clinical Investigation 1992, 90, 1901-1910, 10.1172/jci116067.
- A. Willcox; S. J. Richardson; A. J. Bone; A. K. Foulis; N. G. Morgan; Analysis of islet inflammation in human type 1 diabetes. Clinical & Experimental Immunology 2009, 155, 173-181, 10.1111/j.1365-2249.2008.03860.x.
- Cristiano Scottà; Giorgia Fanelli; Sec Julie Hoong; Marco Romano; Estefania Nova Lamperti; Mitalee Sukthankar; Giuliana Guggino; Henrieta Fazekasova; Kulachelvy Ratnasothy; Pablo Daniel Becker; et al.Behdad AfzaliRobert I. LechlerGiovanna Lombardi Impact of immunosuppressive drugs on the therapeutic efficacy of ex vivo expanded human regulatory T cells. Haematologica 2015, 101, 91-100, 10.3324/haematol.2015.128934.
- David C. Wraith; The Future of Immunotherapy: A 20-Year Perspective. Frontiers in Immunology 2017, 8, 1668-1668, 10.3389/fimmu.2017.01668.
- Nicholas S. Wilson; Dima El-Sukkari; Gabrielle T. Belz; Christopher M. Smith; Raymond J. Steptoe; William R. Heath; Ken Shortman; José A. Villadangos; Most lymphoid organ dendritic cell types are phenotypically and functionally immature. Blood 2003, 102, 2187-2194, 10.1182/blood-2003-02-0513.
- Elodie Segura; Jenny Valladeau-Guilemond; Marie-Hélène Donnadieu; Xavier Sastre-Garau; Vassili Soumelis; Sebastian Amigorena; Characterization of resident and migratory dendritic cells in human lymph nodes. Journal of Experimental Medicine 2012, 209, 653-660, 10.1084/jem.20111457.
- Tomer Granot; Takashi Senda; Dustin J. Carpenter; Nobuhide Matsuoka; Joshua Weiner; Claire L. Gordon; Michelle Miron; Brahma V. Kumar; Adam Griesemer; Siu-Hong Ho; et al.Harvey LernerJoseph J.C. ThomeThomas ConnorsBoris ReizisDonna L. Farber Dendritic Cells Display Subset and Tissue-Specific Maturation Dynamics over Human Life. Immunity 2017, 46, 504-515, 10.1016/j.immuni.2017.02.019.
- Ivan Zanoni; Francesca Granucci; Regulation of antigen uptake, migration, and lifespan of dendritic cell by Toll-like receptors. Journal of Molecular Medicine 2010, 88, 873-880, 10.1007/s00109-010-0638-x.
- F Granucci; C Vizzardelli; E Virzi; M Rescigno; P Ricciardi-Castagnoli; Transcriptional reprogramming of dendritic cells by differentiation stimuli.. European Journal of Immunology 2001, 31, 2-9, 10.1002/1521-4141(200109)31:9<2539::AID-IMMU2539>3.0.CO;2-9.
- Nicholas S. Wilson; Dima El-Sukkari; José A. Villadangos; Dendritic cells constitutively present self antigens in their immature state in vivo and regulate antigen presentation by controlling the rates of MHC class II synthesis and endocytosis. Blood 2004, 103, 2187-2195, 10.1182/blood-2003-08-2729.
- Ralph M. Steinman; Maggie Pack; Kayo Inaba; Dendritic Cell Development and Maturation. Advances in Experimental Medicine and Biology 1997, 417, 1-6, 10.1007/978-1-4757-9966-8_1.
- Jacques Banchereau; Ralph M. Steinman; Dendritic cells and the control of immunity. Nature 1998, 392, 245-252, 10.1038/32588.
- Verena Raker; Matthias Domogalla; Kerstin Steinbrink; Tolerogenic Dendritic Cells for Regulatory T Cell Induction in Man. Frontiers in Immunology 2015, 6, 569, 10.3389/fimmu.2015.00569.
- Caspar Ohnmacht; Andrea Pullner; Susan B.S. King; Ingo Drexler; Stefanie Meier; Thomas Brocker; David Voehringer; Constitutive ablation of dendritic cells breaks self-tolerance of CD4 T cells and results in spontaneous fatal autoimmunity. Journal of Experimental Medicine 2009, 206, 549-559, 10.1084/jem.20082394.
- Shohei Hori; Takashi Nomura; Shimon Sakaguchi; Control of Regulatory T Cell Development by the Transcription Factor Foxp3. Science 2003, 299, 1057-1061, 10.1126/science.1079490.
- Jason D. Fontenot; Marc A. Gavin; Alexander Y. Rudensky; Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nature Immunology 2003, 4, 330-336, 10.1038/ni904.
- James B. Wing; Atsushi Tanaka; Shimon Sakaguchi; Human FOXP3+ Regulatory T Cell Heterogeneity and Function in Autoimmunity and Cancer. Immunity 2019, 50, 302-316, 10.1016/j.immuni.2019.01.020.
- Nicola Gagliani; Chiara F Magnani; Samuel Huber; Monica E Gianolini; Mauro Pala; Paula Licona-Limon; Binggege Guo; De'broski R Herbert; Alessandro Bulfone; Filippo Trentini; et al.Mariaclelia Stefania DI SerioRosa BacchettaMarco AndreaniLeonie BrockmannSilvia GregoriRichard A FlavellMaria-Grazia Roncarolo Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nature Medicine 2013, 19, 739-746, 10.1038/nm.3179.
- Maria Grazia Roncarolo; Silvia Gregori; Rosa Bacchetta; Manuela Battaglia; Nicola Gagliani; The Biology of T Regulatory Type 1 Cells and Their Therapeutic Application in Immune-Mediated Diseases. Immunity 2018, 49, 1004-1019, 10.1016/j.immuni.2018.12.001.
- Mary E Keir; Loise M Francisco; Arlene H Sharpe; PD-1 and its ligands in T-cell immunity. Current Opinion in Immunology 2007, 19, 309-314, 10.1016/j.coi.2007.04.012.
- Juan Wu; Anatolij Horuzsko; Expression and function of immunoglobulin-like transcripts on tolerogenic dendritic cells. Human Immunology 2009, 70, 353-356, 10.1016/j.humimm.2009.01.024.
- D. Brown; J. Trowsdale; Rachel Allen; The LILR family: modulators of innate and adaptive immune pathways in health and disease. Tissue Antigens 2004, 64, 215-225, 10.1111/j.0001-2815.2004.00290.x.
- Ronald H Schwartz; T cell clonal anergy. Current Opinion in Immunology 1997, 9, 351-357, 10.1016/s0952-7915(97)80081-7.
- Gordon J. Freeman; Andrew J. Long; Yoshiko Iwai; Karen Bourque; Tatyana Chernova; Hiroyuki Nishimura; Lori J. Fitz; Nelly Malenkovich; Taku Okazaki; Michael C. Byrne; et al.Heidi F. HortonLynette FouserLaura CarterVincent LingMichael R. BowmanBeatriz M. CarrenoMary CollinsClive R. WoodTasuku Honjo Engagement of the Pd-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. Journal of Experimental Medicine 2000, 192, 1027-1034, 10.1084/jem.192.7.1027.
- Yvette E Latchman; Clive R. Wood; Tatyana Chernova; Divya Chaudhary; Madhuri Borde; Irene Chernova; Yoshiko Iwai; Andrew J. Long; Jacquelyn A Brown; Raquel J Nunes; et al.Edward A. GreenfieldKaren BourqueVassiliki A. BoussiotisLaura L. CarterBeatriz M. CarrenoNelly MalenkovichHiroyuki NishimuraTaku OkazakiTasuku HonjoArlene H. SharpeGordon J. Freeman PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nature Immunology 2001, 2, 261-268, 10.1038/85330.
- Marco Colonna; Francisco Navarro; Teresa Bellón; Manuel Llano; Pilar García; Jacqueline Samaridis; Lena Angman; Marina Cella; Miguel López-Botet; A Common Inhibitory Receptor for Major Histocompatibility Complex Class I Molecules on Human Lymphoid and Myelomonocytic Cells. Journal of Experimental Medicine 1997, 186, 1809-1818, 10.1084/jem.186.11.1809.
- Silvia Gregori; Daniela Tomasoni; Valentina Pacciani; Miriam Scirpoli; Manuela Battaglia; Chiara F Magnani; Ehud Hauben; Maria-Grazia Roncarolo; Differentiation of type 1 T regulatory cells (Tr1) by tolerogenic DC-10 requires the IL-10–dependent ILT4/HLA-G pathway. Blood 2010, 116, 935-944, 10.1182/blood-2009-07-234872.
- Omar S. Qureshi; Yong Zheng; Kyoko Nakamura; Kesley Attridge; Claire Manzotti; Emily M. Schmidt; Jennifer Baker; Louisa E. Jeffery; Satdip Kaur; Zoe Briggs; et al.Tie Z. HouClare FutterGraham AndersonLucy S.K. WalkerDavid M. Sansom Trans-Endocytosis of CD80 and CD86: A Molecular Basis for the Cell-Extrinsic Function of CTLA-4. Science 2011, 332, 600-603, 10.1126/science.1202947.
- Andrew L. Mellor; Derin B. Keskin; Theodore Johnson; Phillip Chandler; David Munn; Cells Expressing Indoleamine 2,3-Dioxygenase Inhibit T Cell Responses. The Journal of Immunology 2002, 168, 3771-3776, 10.4049/jimmunol.168.8.3771.
- Patrick Hwu; Mark X. Du; Réjean Lapointe; My Do; Milton W. Taylor; Howard A. Young; Indoleamine 2,3-Dioxygenase Production by Human Dendritic Cells Results in the Inhibition of T Cell Proliferation. The Journal of Immunology 2000, 164, 3596-3599, 10.4049/jimmunol.164.7.3596.
- Francesca Fallarino; Ursula Grohmann; Sylvaine You; Barbara C. McGrath; Douglas R. Cavener; Carmine Vacca; Ciriana Orabona; Roberta Bianchi; Maria Laura Belladonna; Claudia Volpi; et al.Pere SantamariaMaria C. FiorettiPaolo Puccetti The Combined Effects of Tryptophan Starvation and Tryptophan Catabolites Down-Regulate T Cell Receptor ζ-Chain and Induce a Regulatory Phenotype in Naive T Cells. The Journal of Immunology 2006, 176, 6752-6761, 10.4049/jimmunol.176.11.6752.
- Geon Kook Lee; Hyeon Jin Park; Megan Macleod; Phillip Chandler; David H. Munn; Andrew L. Mellor; Tryptophan deprivation sensitizes activated T cells to apoptosis prior to cell division. Immunology 2002, 107, 452-460, 10.1046/j.1365-2567.2002.01526.x.
- Joshua D. Mezrich; John H. Fechner; Xiaoji Zhang; Brian P. Johnson; William J. Burlingham; Christopher A. Bradfield; An Interaction between Kynurenine and the Aryl Hydrocarbon Receptor Can Generate Regulatory T Cells. The Journal of Immunology 2010, 185, 3190-3198, 10.4049/jimmunol.0903670.
- Ângelo Ferreira Chora; Paulo Fontoura; Andreia Cunha; Teresa Faria Pais; Sílvia Cardoso; Peggy P. Ho; Lowen Y. Lee; Raymond A. Sobel; Lawrence Steinman; Miguel P. Soares; et al. Heme oxygenase–1 and carbon monoxide suppress autoimmune neuroinflammation. Journal of Clinical Investigation 2007, 117, 438-447, 10.1172/jci28844.
- Silvia Gregori; Maria Grazia Roncarolo; Rosa Bacchetta; Methods for In Vitro Generation of Human Type 1 Regulatory T Cells. Advanced Structural Safety Studies 2010, 677, 31-46, 10.1007/978-1-60761-869-0_3.
- Karen Dixon; Sandra W. Van Der Kooij; Dario A. A. Vignali; Cees Van Kooten; Human tolerogenic dendritic cells produce IL-35 in the absence of other IL-12 family members. European Journal of Immunology 2015, 45, 1736-1747, 10.1002/eji.201445217.
- Wanda Niedbala; Xiao-Qing Wei; Beilei Cai; Axel J Hueber; Bernard P. Leung; Iain B. McInnes; Foo Y. Liew; Correction: IL-35 is a novel cytokine with therapeutic effects against collagen-induced arthritis through the expansion of regulatory T cells and suppression of Th17 cells. European Journal of Immunology 2007, 37, 3293-3293, 10.1002/eji.200790047.
- Carolina Obregon; Rajesh Kumar; Manuel Antonio Pascual; Giuseppe Vassalli; Déla Golshayan; Update on Dendritic Cell-Induced Immunological and Clinical Tolerance. Frontiers in Immunology 2017, 8, 1514-1514, 10.3389/fimmu.2017.01514.
- Sheng Xiao; Hulin Jin; Thomas Korn; Sue Min Liu; Mohamed Oukka; Bing Lim; Vijay K. Kuchroo; Retinoic Acid Increases Foxp3+ Regulatory T Cells and Inhibits Development of Th17 Cells by Enhancing TGF-β-Driven Smad3 Signaling and Inhibiting IL-6 and IL-23 Receptor Expression. The Journal of Immunology 2008, 181, 2277-2284, 10.4049/jimmunol.181.4.2277.
- Alfonso Rodríguez Sánchez-Paulete; A. Teijeira; Francisco J. Cueto; S. Garasa; J.L. Pérez-Gracia; Alvaro Sanchez Arraez; David Sancho-Madrid; I. Melero; Antigen cross-presentation and T-cell cross-priming in cancer immunology and immunotherapy. Annals of Oncology 2017, 28, xii44-xii55, 10.1093/annonc/mdx237.
- Neil A. Fanger; Charles R. Maliszewski; Ken Schooley; Thomas S. Griffith; Human Dendritic Cells Mediate Cellular Apoptosis via Tumor Necrosis Factor–Related Apoptosis-Inducing Ligand (Trail). Journal of Experimental Medicine 1999, 190, 1155-1164, 10.1084/jem.190.8.1155.
- Urban Švajger; Primož J. Rožman; Recent discoveries in dendritic cell tolerance-inducing pharmacological molecules. International Immunopharmacology 2020, 81, 106275, 10.1016/j.intimp.2020.106275.
- Jennifer Machen; Jo Harnaha; Robert Lakomy; Alexis Styche; Massimo Trucco; Nick Giannoukakis; Antisense Oligonucleotides Down-Regulating Costimulation Confer Diabetes-Preventive Properties to Nonobese Diabetic Mouse Dendritic Cells. The Journal of Immunology 2004, 173, 4331-4341, 10.4049/jimmunol.173.7.4331.
- Nick Giannoukakis; Brett Phillips; David Finegold; Jo Harnaha; Massimo Trucco; Phase I (Safety) Study of Autologous Tolerogenic Dendritic Cells in Type 1 Diabetic Patients. Diabetes Care 2011, 34, 2026-2032, 10.2337/dc11-0472.
- Lorenzo Piemonti; Paolo Monti; Paola Allavena; M Sironi; L Soldini; Biagio Eugenio Leone; C Socci; V Di Carlo; Glucocorticoids affect human dendritic cell differentiation and maturation.. The Journal of Immunology 1999, 162, 6473-6481.
- Y. Kurochkina; M. Tikhonova; T. Tyrinova; O. Leplina; A. Sizikov; A. Sulutian; O. Chumasova; A. Ostanin; E. Chernykh; SAT0212 The safety and tolerability of intra-articular injection of tolerogenic dendritic cells in patients with rheumatoid arthritis: the preliminary results. Saturday, 16 JUNE 2018 2018, 77, 966-967, 10.1136/annrheumdis-2018-eular.2880.
- Aranzazu Jauregui-Amezaga; Raquel Cabezón; Anna Ramírez-Morros; Carolina España; Jordi Rimola; Concepció Bru; Susana Pinó-Donnay; Marta Gallego; Maria Carme Masamunt; Ingrid Ordas; et al.Miquel LozanoJoan CidJulian PanésDaniel Benítez-RibasElena Ricart Intraperitoneal Administration of Autologous Tolerogenic Dendritic Cells for Refractory Crohn’s Disease: A Phase I Study. Journal of Crohn's and Colitis 2015, 9, 1071-1078, 10.1093/ecco-jcc/jjv144.
- Gabriela Bomfim Ferreira; An-Sofie Vanherwegen; Guy Eelen; Ana Carolina Fierro Gutiérrez; Leentje Van Lommel; Kathleen Marchal; Lieve Verlinden; Annemieke Verstuyf; Tatiane Nogueira; Maria Georgiadou; et al.Frans SchuitDécio L. EizirikConny GysemansPeter CarmelietLut OverberghChantal Mathieu Vitamin D3 Induces Tolerance in Human Dendritic Cells by Activation of Intracellular Metabolic Pathways. Cell Reports 2015, 10, 711-725, 10.1016/j.celrep.2015.01.013.
- Jeremy T. Warshauer; Jeffrey A. Bluestone; Mark S. Anderson; New Frontiers in the Treatment of Type 1 Diabetes. Cell Metabolism 2019, 31, 46-61, 10.1016/j.cmet.2019.11.017.
- G M Bell; Amy Anderson; J Diboll; R Reece; O Eltherington; R A Harry; T Fouweather; C Macdonald; T Chadwick; Elaine McColl; et al.J DunnA M DickinsonCatharien HilkensJohn D Isaacs Autologous tolerogenic dendritic cells for rheumatoid and inflammatory arthritis. Annals of the Rheumatic Diseases 2016, 76, 227-234, 10.1136/annrheumdis-2015-208456.
- Kelan A. Hlavaty; Xunrong Luo; Lonnie D. Shea; Stephen D. Miller; Cellular and molecular targeting for nanotherapeutics in transplantation tolerance. Clinical Immunology 2015, 160, 14-23, 10.1016/j.clim.2015.03.013.
- Sebastian O. Stead; Svjetlana Kireta; Steven James Peter McInnes; Francis D. Kette; Kisha N. Sivanathan; Juewan Kim; Eduardo J. Cueto-Diaz; Frederique Cunin; Jean-Olivier Durand; Christopher J. Drogemuller; et al.Robert P. CarrollNicolas H. VoelckerPatrick T. Coates Murine and Non-Human Primate Dendritic Cell Targeting Nanoparticles for in Vivo Generation of Regulatory T-Cells. ACS Nano 2018, 12, 6637-6647, 10.1021/acsnano.8b01625.
- Vania Manolova; Anna Flace; Monika Bauer; Katrin Schwarz; Philippe Saudan; Martin F. Bachmann; Nanoparticles target distinct dendritic cell populations according to their size. European Journal of Immunology 2008, 38, 1404-1413, 10.1002/eji.200737984.
- Jamal S. Lewis; Natalia V. Dolgova; Ying Zhang; Chang Qing Xia; Clive H. Wasserfall; Mark A. Atkinson; Michael J. Clare-Salzler; Benjamin G. Keselowsky; A combination dual-sized microparticle system modulates dendritic cells and prevents type 1 diabetes in prediabetic NOD mice. Clinical Immunology 2015, 160, 90-102, 10.1016/j.clim.2015.03.023.
- Carl Engman; Yi Wen; Wilson S. Meng; Rita Bottino; Massimo Trucco; Nick Giannoukakis; Generation of antigen-specific Foxp3+ regulatory T-cells in vivo following administration of diabetes-reversing tolerogenic microspheres does not require provision of antigen in the formulation. Clinical Immunology 2015, 160, 103-123, 10.1016/j.clim.2015.03.004.
- Peter A. Gottlieb; Thomas Delong; Rocky L. Baker; Lisa Fitzgerald-Miller; Rebecca Wagner; Gabrielle Cook; Marian R. Rewers; Aaron Michels; Kathryn Haskins; Chromogranin A is a T cell antigen in human type 1 diabetes. Journal of Autoimmunity 2013, 50, 38-41, 10.1016/j.jaut.2013.10.003.
- Suchitra Prasad; Tobias Neef; Dan Xu; Joseph R. Podojil; Daniel R. Getts; Lonnie D. Shea; Stephen D. Miller; Tolerogenic Ag-PLG nanoparticles induce tregs to suppress activated diabetogenic CD4 and CD8 T cells. Journal of Autoimmunity 2017, 89, 112-124, 10.1016/j.jaut.2017.12.010.
- Roberto A. Maldonado; Robert A. LaMothe; Joseph D. Ferrari; Ai-Hong Zhang; Robert Rossi; Pallavi N. Kolte; Aaron P. Griset; Conlin O’Neil; David H. Altreuter; Erica Browning; et al.Lloyd JohnstonOmid FarokhzadRobert LangerDavid W. ScottUlrich H. von AndrianTakashi Kei Kishimoto Polymeric synthetic nanoparticles for the induction of antigen-specific immunological tolerance. Proceedings of the National Academy of Sciences 2014, 112, E156-E165, 10.1073/pnas.1408686111.
- Giuseppe Cappellano; Abiy Demeke Woldetsadik; Elisabetta Orilieri; Yogesh Shivakumar; Manuela Rizzi; Fabio Carniato; Casimiro Luca Gigliotti; Elena Boggio; Nausicaa Clemente; Cristoforo Comi; et al.Chiara DianzaniRenzo Luciano BoldoriniAnnalisa ChiocchettiFilippo RenòUmberto Dianzani Subcutaneous inverse vaccination with PLGA particles loaded with a MOG peptide and IL-10 decreases the severity of experimental autoimmune encephalomyelitis. Vaccine 2014, 32, 5681-5689, 10.1016/j.vaccine.2014.08.016.
- Tobias L. Freitag; Joseph R. Podojil; Ryan M. Pearson; Frank J. Fokta; Cecilia Sahl; Marcel Messing; Leif C. Andersson; Katarzyna Leskinen; Päivi Saavalainen; Lisa I. Hoover; et al.Kelly HuangDeborah PhippardSanaz MalekiNicholas J.C. KingLonnie D. SheaStephen D. MillerSeppo K. MeriDaniel R. Getts Gliadin Nanoparticles Induce Immune Tolerance to Gliadin in Mouse Models of Celiac Disease. Gastroenterology 2020, 158, 1667-1681.e12, 10.1053/j.gastro.2020.01.045.
- Ciarán P. Kelly; Joseph A. Murray; Daniel A. Leffler; Daniel R. Getts; Adam C. Bledsoe; Glennda Smithson; M. Roy First; Amy Morris; Michael Boyne; Adam Elhofy; et al.Tsung-Teh WuJoseph R. PodojilStephen D. MillerRobert FogelTobias L. FreitagMichele GerberPaul K. HaynesMichael KorenMark MatsonSeppo MeriThomas H. OliphantBarbara E. RizzardiJocelyn SilvesterMark Turner TAK-101 Nanoparticles Induce Gluten-Specific Tolerance in Celiac Disease: A Randomized, Double-Blind, Placebo-Controlled Study. Gastroenterology 2021, 161, 66-80.e8, 10.1053/j.gastro.2021.03.014.
- Tolerogenic Immunotherapy: Targeting DC Surface Receptors to Induce Antigen-Specific Tolerance . frontiersin. Retrieved 2021-10-8
- Courtney A. Iberg; Daniel Hawiger; Targeting Dendritic Cells with Antigen-Delivering Antibodies for Amelioration of Autoimmunity in Animal Models of Multiple Sclerosis and Other Autoimmune Diseases. Antibodies 2020, 9, 23, 10.3390/antib9020023.
- Andrew Jones; Jessica Bourque; Lindsey Kuehm; Adeleye Opejin; Ryan M. Teague; Cindy Gross; Daniel Hawiger; Immunomodulatory Functions of BTLA and HVEM Govern Induction of Extrathymic Regulatory T Cells and Tolerance by Dendritic Cells. Immunity 2016, 45, 1066-1077, 10.1016/j.immuni.2016.10.008.
- Daniel Hawiger; Kayo Inaba; Yair Dorsett; Ming Guo; Karsten Mahnke; Miguel Rivera; Jeffrey V. Ravetch; Ralph M. Steinman; Michel C. Nussenzweig; Dendritic Cells Induce Peripheral T Cell Unresponsiveness under Steady State Conditions in Vivo. Journal of Experimental Medicine 2001, 194, 769-780, 10.1084/jem.194.6.769.
- Joel N. H. Stern; Derin B. Keskin; Zenichiro Kato; Hanspeter Waldner; Sonja Schallenberg; Ana Anderson; Harald von Boehmer; Karsten Kretschmer; Jack L. Strominger; Promoting tolerance to proteolipid protein-induced experimental autoimmune encephalomyelitis through targeting dendritic cells. Proceedings of the National Academy of Sciences 2010, 107, 17280-17285, 10.1073/pnas.1010263107.
- Daniel Hawiger; Revati F Masilamani; Estelle Bettelli; Vijay K Kuchroo; Michel C Nussenzweig; Immunological Unresponsiveness Characterized by Increased Expression of CD5 on Peripheral T Cells Induced by Dendritic Cells In Vivo. Immunity 2004, 20, 695-705, 10.1016/j.immuni.2004.05.002.
- Rachel Spiering; Bram Margry; Chantal Keijzer; Cathleen Petzold; Aad Hoek; Josée Wagenaar-Hilbers; Ruurd van der Zee; Willem van Eden; Karsten Kretschmer; Femke Broere; et al. DEC205+ Dendritic Cell–Targeted Tolerogenic Vaccination Promotes Immune Tolerance in Experimental Autoimmune Arthritis. The Journal of Immunology 2015, 194, 4804-4813, 10.4049/jimmunol.1400986.
- Munisch Wadwa; Robert Klopfleisch; Jan Buer; Astrid M. Westendorf; Targeting antigens to DEC-205 on dendritic cells induces immune protection in experimental colitis in mice. European Journal of Microbiology and Immunology 2016, 6, 1-8, 10.1556/1886.2015.00048.
- Olivier P. Joffre; David Sancho; Santiago Zelenay; Anna M. Keller; Caetano Reis e Sousa; Efficient and versatile manipulation of the peripheral CD4 + T‐cell compartment by antigen targeting to DNGR‐1/CLEC9A. European Journal of Immunology 2010, 40, 1255-1265, 10.1002/eji.201040419.
- Julie Wang; Hugh A. Sampson; Safety and efficacy of epicutaneous immunotherapy for food allergy. Pediatric Allergy and Immunology 2018, 29, 341-349, 10.1111/pai.12869.
- Vincent Dioszeghy; Lucie Mondoulet; Léo Laoubi; Véronique Dhelft; Camille Plaquet; Adeline Bouzereau; Christophe Dupont; Hugh Sampson; Antigen Uptake by Langerhans Cells Is Required for the Induction of Regulatory T Cells and the Acquisition of Tolerance During Epicutaneous Immunotherapy in OVA-Sensitized Mice. Frontiers in Immunology 2018, 9, 1951, 10.3389/fimmu.2018.01951.
- Monika Tutaj; Marian Szczepanik; Epicutaneous (EC) immunization with myelin basic protein (MBP) induces TCRαβ+ CD4+ CD8+ double positive suppressor cells that protect from experimental autoimmune encephalomyelitis (EAE). Journal of Autoimmunity 2007, 28, 208-215, 10.1016/j.jaut.2007.02.017.
- Maciej Jurynczyk; Agata Walczak; Anna Jurewicz; Dorota Jesionek-Kupnicka; Marian Szczepanik; Krzysztof Selmaj; Immune regulation of multiple sclerosis by transdermally applied myelin peptides. Annals of Neurology 2010, 68, 593-601, 10.1002/ana.22219.
- Agata Walczak; Malgorzata Siger; Agnieszka Ciach; Marian Szczepanik; Krzysztof Selmaj; Transdermal Application of Myelin Peptides in Multiple Sclerosis Treatment. JAMA Neurology 2013, 70, 1105-1109, 10.1001/jamaneurol.2013.3022.
- Monika Majewska-Szczepanik; Marta Góralska; Katarzyna Marcińska; Magdalena Zemelka-Wiącek; Anna Strzępa; Iwona Dorożyńska; Marian Szczepanik; Epicutaneous immunization with protein antigen TNP-Ig alleviates TNBS-induced colitis in mice. Pharmacological Reports 2012, 64, 1497-1504, 10.1016/s1734-1140(12)70947-7.
- Katarzyna Marcińska; Monika Majewska-Szczepanik; Agata Lazar; Paulina Kowalczyk; Dominika Biała; Dorota Woźniak; Marian Szczepanik; Epicutaneous (EC) immunization with type II collagen (COLL II) induces CD4 + CD8 + T suppressor cells that protect from collagen-induced arthritis (CIA). Pharmacological Reports 2015, 68, 483-489, 10.1016/j.pharep.2015.11.004.
- Anja Ten Brinke; Marc Martinez-Llordella; Nathalie Cools; Catharien M. U. Hilkens; S. Marieke van Ham; Birgit Sawitzki; Edward K. Geissler; Giovanna Lombardi; Piotr Trzonkowski; Eva Martinez-Caceres; et al. Ways Forward for Tolerance-Inducing Cellular Therapies- an AFACTT Perspective. Frontiers in Immunology 2019, 10, 181, 10.3389/fimmu.2019.00181.
- Matthias P. Domogalla; Patricia V. Rostan; Verena Raker; Kerstin Steinbrink; Tolerance through Education: How Tolerogenic Dendritic Cells Shape Immunity. Frontiers in Immunology 2017, 8, 1764, 10.3389/fimmu.2017.01764.
- Bechara Mfarrej; Eleonora Tresoldi; Angela Stabilini; Alessia Paganelli; Rossana Caldara; Antonio Secchi; Manuela Battaglia; Generation of donor-specific Tr1 cells to be used after kidney transplantation and definition of the timing of their in vivo infusion in the presence of immunosuppression. Journal of Translational Medicine 2017, 15, 40, 10.1186/s12967-017-1133-8.
- Bechara Mfarrej; Eleonora Tresoldi; Angela Stabilini; Alessia Paganelli; Rossana Caldara; Antonio Secchi; Manuela Battaglia; Generation of donor-specific Tr1 cells to be used after kidney transplantation and definition of the timing of their in vivo infusion in the presence of immunosuppression. Journal of Translational Medicine 2017, 15, 40, 10.1186/s12967-017-1133-8.