Multiple sclerosis (MS) is an autoimmune disease of the Central Nervous System, characterized by an inflammatory process leading to the destruction of myelin with neuronal death and neurodegeneration. In MS, lymphocytes cross the blood-brain barrier, creating inflammatory demyelinated plaques located primarily in the white matter. MS potential treatments involve various mechanisms of action on immune cells, immunosuppression, inhibition of the passage through the blood-brain barrier, and immunotolerance.
- inflammation
- nanotechnology
- immunotolerance
- drug delivery system
1. Introduction
Multiple sclerosis (MS) is a chronic inflammatory disease of the Central Nervous System (CNS) [1,2][1][2], caused by a response of T and B cells towards the myelin sheath surrounding the axons of the CNS [3,4][3][4]. This process triggers an early inflammatory state, which ultimately culminates in demyelination and neuronal degeneration [2,5][2][5]. The origin of MS is still unknown. It is assumed that genetic and environmental factors (smoking, toxins), low vitamin D levels, obesity, microbial and viral infections, can contribute to its onset [1]. Worldwide, 2.3 million people are affected by MS, with an incidence rate of approximately 50–300 per 100,000 people [6]. MS can evolve into different profiles, Relapse-Remitting (RR-MS), in most patients (80–85% of MS cases) and, less common, primary progressive course (PP-MS) [7]. RR-MS can turn into a progressive course, referred to as secondary progressive (SP-MS) [7]. The typical neurological symptoms of MS include visual field disturbance, double and blurred vision, motor symptoms such as weakness of the face and extremities, bladder/bowel problems, walking difficulties, spasticity (stiffness and muscle spasms), and dizziness. The diagnosis of multiple sclerosis is based on the integration of clinical, imaging, and laboratory findings [7].
Treatment “first-line therapies” are mainly aimed at preventing the onset or relapse of patients. The administration of anti-inflammatory drugs is the most common therapy; attempts are made to extensively suppress the immune system, leading to the deletion or inactivation of entire subgroups of T cells [8].
In addition, to MS immunosuppressive treatments, there are other mechanisms of action on immune cells aimed to inhibit their passage through the Blood-Brain Barrier (BBB) and their immunotolerance. Interferon (IFN) administration, as IFN beta-1α and IFN beta-1β, has generally been used as “first-line therapy” [8], injected either intramuscularly or subcutaneously. IFN-beta 1α and 1β modulate T and B cells and regulate cytokine release [9]; however, these treatments exhibit a series of adverse effects and are only moderately efficacious. Natalizumab (the first approved intravenous drug) is a monoclonal antibody against integrin α4, which blocks the entry of lymphocytes into the CNS [10]. Although the efficiency of Natalizumab in MS relapse is superior to that of IFN, its administration is accompanied by noxious side effects [10]. Therefore, it is used in MS patients who do not respond positively to other treatments. Rituximab and Obinutuzumabis are antibodies against CD20 on B-lymphocytes, causing their depletion. Mitoxantrone has cytotoxic effects against B cells as well as T-helper and T-cytotoxic lymphocytes, but its use is limited due the side effects. In addition, several approaches aimed at restoring immune tolerance were tested [11]. Indeed, it is thought that the first event occurring in the MS pathogenesis is a tolerance breakdown, which determines the activation of naive myelin-specific T cells in healthy individuals [11]. Antigen-specific immunotherapies aim to restore immune tolerance without suppressing overall immune surveillance against microbes and cancer [11].
2. Why Is a Nano-Therapeutic Approach Needed to Treat MS?
Multiple sclerosis is a complex disease, and its underlying mechanisms are only partially understood. Moreover, the involvement not only of peripheral immune cells (T and B lymphocytes) but also of the central ones (microglia) makes the therapeutic target complex. Therefore, in addition to acting at the peripheral level, the treatment must also act at the level of the CNS. Here, MS therapy is accompanied by low efficacy due to the presence of BBB and the occurrence of side effects due to the dispersion of the drugs that fail to enter the CNS [13][12]. Furthermore, it is important to administer therapeutic agents to the MS lesions present in different place of the CNS, without affecting other normal CNS tissues in order to avoid further damages. The use of nanoscale materials is expected to provide unique opportunities to: (i) improve drug solubility and bioavailability; (ii) enable targeted delivery and controlled release and, consequently, (iii) increase effective routes of administration and (iv) reduce toxicity. Interestingly, not only can nanovectors (extracellular and artificial vesicle nanoparticles) act as carriers of relevant molecules, but they can also trigger an immunomodulatory effect [13][12]. Indeed, variations in the chemical composition, size, and shape of the nanovector have a different impact on the immune target and immune response that could be even more significant in the context of autoimmune diseases [13][12]. In MS, the use of nanovectors has been investigated as drug delivery systems and as vectors for antigen-specific immunomodulation.
3. Extracellular Vesicles
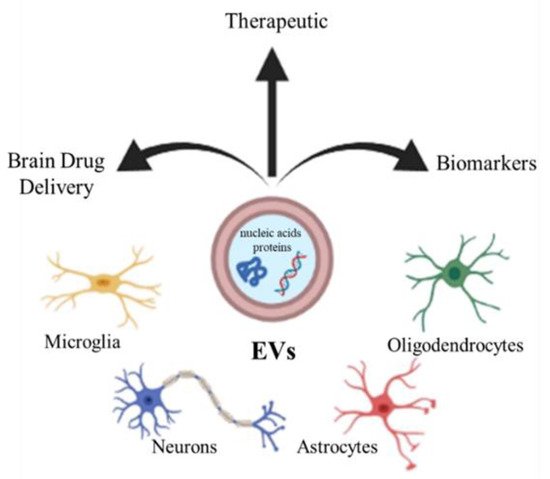
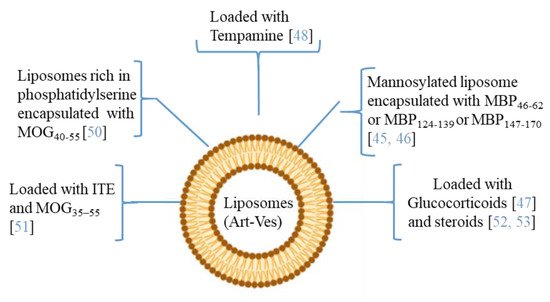
4. Artificial Vesicles
References
- Amato, M.P.; Ponziani, G.; Bartolozzi, M.L.; Siracusa, G. A prospective study on the natural history of multiple sclerosis: Clues to the conduct and interpretation of clinical trials. J. Neurol. Sci. 1999, 168, 96106.
- Lassmann, H. Multiple Sclerosis Pathology. Cold Spring Harbor Perspect. Med. 2018, 8, a028936.
- Weber, M.S.; Hemmer, B. Cooperation of B cells and T cells in the pathogenesis of multiple sclerosis. Res. Probl. Cell Differ. 2010, 51, 115–126.
- Van Langelaar, J.; Rijvers, L.; Smolders, J.; Van Luijn, M.M. B and T Cells Driving Multiple Sclerosis: Identity, Mechanisms and Potential Triggers. Front. Immunol. 2020, 11, 760.
- Stadelmann, C.; Wegner, C.; Brück, W. Inflammation, demyelination, and degeneration recent insights from MS pathology. Biochim. Biophys. Acta 2011, 1812, 275–282.
- Thompson, A.J.; Baranzini, S.E.; Geurts, J.; Hemmer, B.; Ciccarelli, O. Multiple sclerosis. Lancet 2018, 21, 1622–1636.
- Tsang, B.K.-T.; Macdonell, R. Multiple sclerosis diagnosis, management and prognosis. Aust. Fam. Physician 2011, 40, 948–955.
- Dhib-Jalbut, S.; Marks, S. Interferon-b mechanisms of action in multiple sclerosis. Neurology 2010, 74, S17–S24.
- Reuss, R. PEGylated interferon beta-1a in the treatment of multiple sclerosis—An update. Biol. Targets Ther. 2013, 7, 131.
- Chataway, J.; Miller, D.H. Natalizumab therapy for multiple sclerosis. Neurotherapeutics 2013, 10, 19–28.
- Goverman, J.M. Immune tolerance in multiple sclerosis. Immunol. Rev. 2011, 241, 228–240.
- Zhang, Q.; Dai, X.; Zhang, H.; Zeng, Y.; Luo, K.; Li, W. Recent advances in development of nanomedicines for multiple sclerosis diagnosis. Biomed. Mater. 2021, 16, 024101.
- Cocucci, E.; Racchetti, G.; Meldolesi, J. Shedding microvesicles: Artefacts no more. Trends Cell Biol. 2009, 19, 43–51.
- Porro, C.; Trotta, T.; Panaro, M.A. Microvesicles in the brain: Biomarker, messenger or mediator? J. Neuroimmunol. 2015, 288, 70–78.
- Yáñez-Mó, M.; Siljander, P.R.-M.; Andreu, Z.; Bedina Zavec, A.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066.
- Fiandaca, M.S.; Kapogiannis, D.; Mapstone, M.; Boxer, A.; Eitan, E.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Federoff, H.J.; Miller, B.L.; et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimer’s Dement. 2015, 11, 600–607.
- Goetzl, E.J.; Kapogiannis, D.; Schwartz, J.B.; Lobach, I.V.; Goetzl, L.; Abner, E.L.; Jicha, G.A.; Karydas, A.M.; Boxer, A.; Miller, B.L. Decreased synaptic proteins in neuronal exosomes of frontotemporal dementia and Alzheimer’s disease. FASEB J. 2016, 30, 4141–4148.
- Tkach, M.; Théry, C. Communication by extracellular vesicles: Where we are and where we need to go. Cell 2016, 164, 1226–1232.
- Raposo, G.; Stahl, P.D. Extracellular vesicles: A new communication paradigm? Nat. Rev. Mol. Cell Biol. 2019, 20, 509–510.
- Russell, A.E.; Sneider, A.; Witwer, K.W.; Bergese, P.; Bhattacharyya, S.N.; Cocks, A.; Cocucci, E.; Erdbrügger, U.; Falcon-Perez, J.M.; Freeman, D.W.; et al. Biological membranes in EV biogenesis, stability, up-take, and cargo transfer: An ISEV position paper arising from the ISEV membranes and EVs workshop. J. Extracell. Vesicles 2019, 8, 1684862.
- Lai, C.P.; Breakefield, X.O. Role of exosomes/microvesicles in the nervous system and use in emerging therapies. Front. Physiol. 2012, 3, 228.
- Dolcetti, E.; Bruno, A.; Guadalupi, L.; Rizzo, F.R.; Musella, A.; Gentile, A.; De Vito, F.; Caioli, S.; Bullitta, S.; Fresegna, D.; et al. Emerging Role of Extracellular Vesicles in the Pathophysiology of Multiple Sclerosis. Int. J. Mol. Sci. 2020, 21, 7336.
- Croese, T.; Furlan, R. Extracellular vesicles in neurodegenerative diseases. Mol. Asp. Med. 2018, 60, 52–61.
- Laso-Garcìa, F.; Ramos-Cejudo, J.; Carrillo-Salinas, F.J.; Otero-Ortega, L.; Feliú, A.; Gómez-de Frutos, M.; Mecha, M.; Díez-Tejedor, E.; Guaza, C.; Gutiérrez-Fernández, M. Therapeutic potential of extracellular vesicles derived from human mesenchymal stem cells in a model of progressive multiple sclerosis. PLoS ONE 2018, 13, e0202590.
- Murphy, A.C.; Lalor, S.J.; Lynch, M.A.; Mills, K.H. Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain Behav. Immun. 2010, 24, 641–651.
- Lombardi, M.; Parolisi, R.; Scaroni, F.; Bonfanti, E.; Gualerzi, A.; Gabrielli, M.; Kerlero de Rosbo, N.; Uccelli, A.; Giussani, P.; Viani, P.; et al. Detrimental and protective action of microglial extracellular vesicles on myelin lesions: Astrocyte involvement in remyelination failure. Acta Neuropathol. 2019, 138, 987–1012.
- Pusic, A.D.; Pusic, K.M.; Clayton, B.L.; Kraig, R.P. IFNg-stimulated dendritic cell exosomes as a potential therapeutic for remyelination. J. Neuroimmunol. 2014, 266, 12–23.
- Casella, G.; Colombo, F.; Finardi, A.; Descamps, H.; Ill-Raga, G.; Spinelli, A.; Podini, P.; Bastoni, M.; Martino, G.; Muzio, L.; et al. Extracellular vesicles containing IL-4 modulate neuroinflammation in a mouse model of multiple sclerosis. Mol. Ther. 2018, 26, 2107–2118.
- Leggio, L.; Arrabito, G.; Ferrara, V.; Vivarelli, S.; Paternò, G.; Marchetti, B.; Pignataro, B.; Iraci, N. Mastering the Tools: Natural versus Artificial Vesicles in Nanomedicine. Adv. Healthc. Mater. 2020, 9, 2000731.
- Hwang, J.Y.; Li, Z.; Loh, X.J. Small molecule therapeutic loaded liposomes as therapeutic carriers: From development to clinical applications. RSC Adv. 2016, 6, 70592–70615.
- Yuan, D.F.; Zong, T.L.; Gao, H.L.; He, Q. Cell penetrating peptide TAT and brain tumor targeting peptide T7 dual modified liposome preparation and in vitro targeting evaluation. Yao Xue Xue Bao 2015, 50, 104–110.
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal formulations in clinical use: An updated review. Pharmaceutics 2017, 9, 12.
- Lutterotti, A.; Hayward-Koennecke, H.; Sospedra, M.; Martin, R. Antigen-Specific Immune Tolerance in Multiple Sclerosis-Promising Approaches and How to Bring Them to Patients. Front. Immunol. 2021, 22, 640935.
- Zeng, H.; Zhang, R.; Jin, B.; Chen, L. Type 1 regulatory T cells: A new mechanism of peripheral immune tolerance. Cell. Mol. Immunol. 2015, 12, 566–571.
- Belogurov, A.A.; Stepanov, A.V.; Smirnov, I.V.; Melamed, D.; Bacon, A.; Mamedov, A.E.; Boitsov, V.M.; Sashchenko, L.P.; Ponomarenko, N.A.; Sharanova, S.N.; et al. Liposome-encapsulated peptides protect against experimental allergic encephalitis. FASEB J. 2013, 27, 222–231.
- Ivanova, V.V.; Khaiboullina, S.F.; Gomzikova, M.O.; Martynova, E.V.; Ferreira, A.M.; Garanina, E.E.; Sakhapov, D.I.; Lomakin, Y.A.; Khaibullin, T.I.; Granatov, E.V.; et al. Divergent Immunomodulation Capacity of Individual Myelin Peptides—Components of Liposomal Therapeutic against Multiple Sclerosis. Front. Immunol. 2017, 8, 1335.
- Avnir, Y.; Turjeman, K.; Tulchinsky, D.; Sigal, A.; Kizelsztein, P.; Tzemach, D.; Gabizon, A.; Barenholz, Y. Fabrication principles and their contribution to the superior in vivo therapeutic efficacy of nano-liposomes remote loaded with glucocorticoids. PLoS ONE 2011, 6, e25721.
- Kizelsztein, P.; Ovadia, H.; Garbuzenko, O.; Sigal, A.; Barenholz, Y. Pegylated nanoliposomes remote-loaded with the antioxidant tempamine ameliorate experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2009, 213, 20–25.
- Jain, A.; Jain, A.; Gulbake, A.; Shilpi, S.; Hurkat, P.; Jain, S.K. Peptide and protein delivery using new drug delivery systems. Crit. Rev TM. Ther. Drug Carrier Syst. 2013, 30, 293–329.
- Pujol-Autonell, I.; Mansilla, M.J.; Rodriguez-Fernandez, S.; Cano-Sarabia, M.; Navarro-Barriuso, J.; Ampudia, R.M.; Rius, A.; Garcia-Jimeno, S.; Perna-Barrull, D.; Martinez-Caceres, E.; et al. Liposome-based immunotherapy against autoimmune diseases: Therapeutic effect on multiple sclerosis. Nanomedicine 2017, 12, 1231–1242.
- Kenison, J.E.; Jhaveri, A.; Li, Z.; Khadse, N.; Tjon, E.; Tezza, S.; Nowakowska, D.; Plasencia, A.; Stanton, V.P., Jr.; Sherr, D.H.; et al. Tolerogenic nanoparticles suppress central nervous system inflammation. Proc. Natl. Acad. Sci. USA 2020, 15, 32017–32028.
- Schmidt, J.; Metselaar, J.M.; Wauben, M.H.M.; Toyka, K.V.; Storm, G.; Gold, R. Long-Circulating Drug Targeting by Liposomal Glucocorticosteroids Increases Therapeutic Efficacy in a Model of Multiple Sclerosis. Brain 2003, 126, 1895–1904.
- Linker, R.A.; Weller, C.; Lühder, F.; Mohr, A.; Schmidt, J.; Knauth, M.; Metselaar, J.M.; Gold, R. Liposomal Glucocorticosteroids in Treatment of Chronic Autoimmune Demyelination: Long-Term Protective effects and Enhanced Efficacy of Methylprednisolone Formulations. Exp. Neurol. 2008, 211, 397–406.