Dapsone (DDS) is an antibacterial drug with well-known antioxidant properties. However, the antioxidant behavior of its derivatives has not been well explored. In the present work, the antioxidant activity of 10 dapsone derivatives 4-substituted was determined by an evaluation in two in vitro models (DPPH radical scavenging assay and ferric reducing antioxidant power). These imine derivatives 1–10 were obtained through condensation between DDS and the corresponding aromatic aldehydes 4-substuited. Three derivatives presented better results than DDS in the determination of DPPH (2, 9, and 10). Likewise, we have three compounds with better reducing activity than dapsone (4, 9, and 10). In order to be more insight, the redox process, a conceptual DFT analysis was carried out. Molecular descriptors such as electronic distribution, the total charge accepting/donating capacity (I/A), and the partial charge accepting/donating capacity (ω+/ω−) were calculated to analyze the relative donor-acceptor capacity through employing a donor acceptor map (DAM). The DFT calculation allowed us to establish a relationship between GAPHOMO-LUMO and DAM with the observed antioxidant effects. According to the results, we concluded that compounds 2 and 3 have the lowest Ra values, representing a good antioxidant behavior observed experimentally in DPPH radical capturing. On the other hand, derivatives 4, 9, and 10 display the best reducing capacity activity with the highest ω− and Rd values. Consequently, we propose these compounds as the best antireductants in our DDS imine derivative series.
1. Introduction
Dapsone (4,4′-diaminodiphenylsulfone, DDS) is an aniline derivative that belongs to the sulfone drug class. It is mainly used for leprosy treatment in combination with rifampicin and clofazimine
[1][2][1,2]. Additionally, DDS has shown a prophylactic effect against
T. gondii and
P. jiroveci on clinical trials with HIV patients
[3][4][3,4]. Furthermore, an antiparasitic effect against
P. falciparum has been reported with DDS in combination with chlorproguanil
[5]. Once absorbed, DDS is metabolized into monoacetyldapsone and
N-hydroxyldapsone
[6], being the one oxidation product responsible for the methemoglobinemia
[7][8][9][7,8,9].
Moreover, DDS possesses an anti-inflammatory effect similar to other non-steroidal anti-inflammatory drugs
[10]. It is a consequence of scavenging for oxidizing species, such as superoxide dismutase, peroxidase, and compounds such as β-carotene
[11]. This activity has been explored in neuroscience research, where DDS diminishes the damage caused by oxidative stress in murine models
[12].
The antioxidant properties of DDS have been explored as a corrosion inhibitor on steel imbibed on the acid medium (HCl 1 M and H
2SO
4 0.5 M), showing an anticorrosive activity > 90% at DDS 400 ppm
[13]. DDS imine derivatives with salicylaldehyde
[14], indol-3-carboxaldehyde, thiophene-2-carboxaldehyde
[15], and benzaldehyde
[16] have shown the anticorrosive effect at the same level as the parent drug.
A structural modification strategy for drugs containing the amine functionality consists of derivatizing into amides or imines. The imine group has the advantage, from the synthetic point, of being easily accessible, which can improve the solubility and biological activity compared to the precursor amines
[17][18][17,18]. Derivatization also positively impacts DDS solubility (0.16 mg/mL) when it reacts with aliphatic amino acids to reach aqueous solubilities higher than 25 mg/mL
[19]. Additionally, the derivatives reported by Wadher et al., five derivatives of 4-substituted DDS imines maintain their antibacterial and antifungal activity against
E. coli,
S. aureus,
A. niger, and
C. albicans [20]. In recent years, the use of computational methods has increased considerably since they allow us to understand and explain various structural and electronic properties of molecules and correlate them with experimental data
[21]. The calculation of quantum molecular descriptors is a useful tool in the study of chemical structures with antioxidant activity. The highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital (LUMO), the ionization potential (I), and electronic affinity (A) are helpful to understand the molecular stability linked with the ability to accept or donate electrons
[22][23][24][22,23,24]. The energy values of HOMO, LUMO, and I have also been associated with the amine groups’ reactivity and toxicity of DDS. Therefore, the amine group is the highest nucleophilic site and susceptible to oxidization
[25]. These parameters will allow the generation of the donor-acceptor map (DAM), a useful tool to categorize any potential antioxidant substance as an electron donor or acceptor that permits us to better understand the possible antioxidant mechanism
[26].
This work aims to: (i) Obtain new aromatic imine derivatives of DDS with aromatic aldehydes substituted in position 4, in order to evaluate their in vitro antioxidant activity through a chemical method using two experimental methodologies; and (ii) establish their correlationship with electronic descriptors of DFT calculations.
The selection of 4-substituted derivatives in this research project was based on literature reports where the antimicrobial and antioxidant activity of imine-type derivatives have a greater effect if the substituents occupy that position
[27][28][27,28]. Furthermore, our study was directed in a complementary way to evaluate the influence of the type of electron-withdrawing/electron-attracting substituents in that position in DDS derivatives.
2. Development and Findings
We prepared and characterized a DDS imine derivative series to assess their antioxidant potential compared to the parental drug. This compound series was evaluated by two in vitro models: (i) DPPH radical capturing; and (ii) ferric reducing antioxidant power. Our experimental findings were complemented with the DFT analysis in terms of (i) electronic distribution; (ii) the total charge accepting/donating capacity (I/A); (iii) the partial charge accepting/donating capacity (ω
+/ω
−); and (iv) an analysis of the relative donor-acceptor capacity through the DAM.
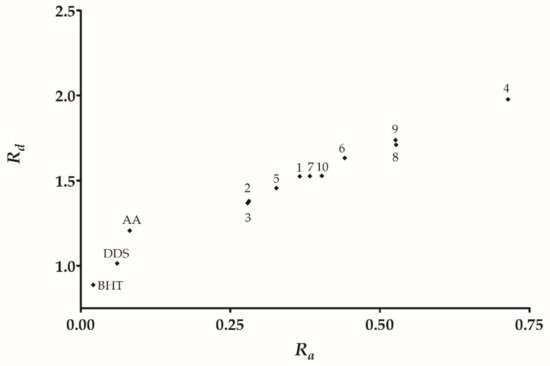
From the DPPH model, we found three trends: (a) Derivatives with null or lower activity than DDS, this set of derivatives have electron withdrawal groups (halogen
5–
7, cya-no
8, and hydrogen
1); (b) derivatives with an equivalent activity to DDS, hydroxy
2, and cinnamyl
10; and (c) derivatives with higher activity than DDS, specifically derivative
9 containing a carboxyl group. Therefore, the following tendency is observed carboxyl >> hydroxyl > cinnamyl > methoxy. Additionally, the reducing antioxidant/power showed a similar tendency, with the derivatives being minor or of similar activity to DDS that contain electron withdrawal groups. Moreover, the higher reducing power derivatives are carboxyl > nitro > cinnamyl > methoxy.
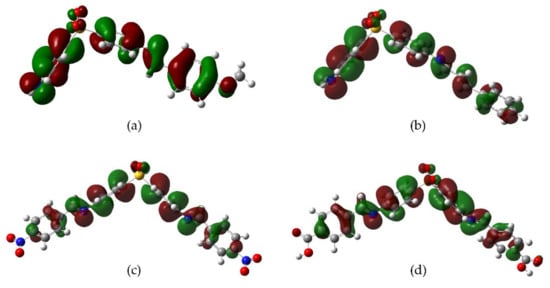
The antioxidant phenomena observed can be explained by an electronic behavior analysis. For derivatives
2,
3, and
10, the substituents act as electron donators (good antiradical agents) due to the induction or hyperconjugation that makes it capable of stabilizing the cation-radical species formed in the electron-donating process towards DPPH or Fe
3+ (
Figure 13). Derivative
9 is also capable of stabilizing the cation-radical due to the electronic resonance on the carboxyl group. In the case of
4, the resonance by the nitro group explains its effectivity in the reducing power model. On the contrary, those derivatives with electron-withdrawing groups (F, Cl, Br, CN) decrease the electronic transfer availability to DPPH radical. Consequently, a null or minor activity with respect to DDS is observed.
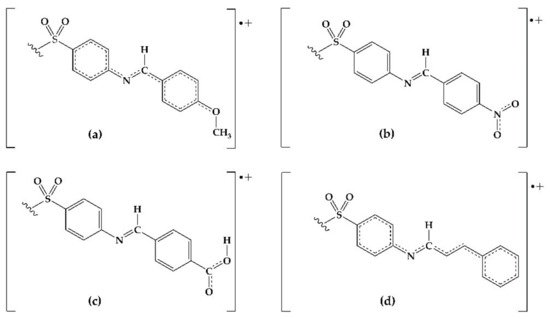
Figure 13. Possible resonant structures of derivatives (a) 2, (b) 4, (c) 9, and (d) 10.
The calculation of molecular electronic parameters is relevant to predict the reactivity in several chemical reactions. The frontier molecular orbitals (HOMO and LUMO) are two crucial descriptors since they represent the electron-donating and electron-withdrawing capacities of a given molecule. Comparing the HOMO values of DDS and its derivatives (
Table 14), we observed that all of the compounds presented lower values than DDS. Among the derivative series,
2,
3, and
10 have higher HOMO values that can be understood as a greater facility towards the electron donation, explaining the suitable antioxidant activities observed in vitro. On the other hand, according to the Gap
HOMO-LUMO, derivatives
4,
8, and
10 have smaller values that involve a higher reactivity correlated to the antioxidant behavior in reducing the power assay (
Table 23).
Table 1. HOMO, LUMO, and GapHOMO-LUMO energy values.
| Derivative |
HOMO (eV) |
LUMO (eV) |
GapHOMO-LUMO (eV) |
| 1 |
The low I values represent compounds that are easily oxidizable and act as efficient antiradical agents depending on their specific electro-donative capacity. For example, in
Table 35, we found that derivatives
2,
3,
5, and
10 have the lowest I value that can be understood as a higher ease to transfer one electron and, therefore have better antiradical activity. In opposition, derivatives
1,
4,
6, and
9 have the highest I value in the series, explaining the low DPPH radical capturing activity of these compounds, except for compound
9.
Table 3. Molecular descriptors calculated for DDS and its derivatives at a CAM-B3LYP theory level and 6-311G (d,p) basis set. Values in eV.
| Compound |
I |
A |
ω− |
ω+ |
Ra |
Rd |
| −7.82 |
−0.99 |
6.83 |
| 1 |
8.14 |
0.64 |
5.24 |
0.84 |
0.37 |
1.53 |
2 |
−7.42 |
−0.69 |
6.72 |
| 2 |
7.84 |
0.33 |
4.73 |
0.65 |
0.28 |
1.38 |
3 |
−7.37 |
| 3 |
7.77−1.93 |
0.336.70 |
| 4.69 |
0.64 |
0.28 |
1.37 |
4 |
−8.33 |
−2.03 |
6.31 |
| 4 |
8.63 |
1.65 |
6.79 |
1.65 |
0.71 |
1.98 |
5 |
−7.52 |
−0.89 |
6.63 |
| 5 |
7.98 |
0.51 |
5.00 |
0.75 |
0.33 |
6 |
−7.96 |
−1.23 |
6.73 |
| 7 |
−7.54 |
−1.05 |
6.49 |
| 8 |
−7.66 |
−1.57 |
6.09 |
| 9 |
−8.05 |
−1.50 |
6.55 |
| 10 |
−7.43 |
−1.15 |
6.28 |
| DDS |
−7.25 |
−0.30 |
6.95 |
| BHT |
−7.17 |
1.23 |
8.40 |
| AA |
−8.22 |
0.42 |
8.64 |
Table 2. Percentage of the reducing effect and correlation with DDS and ascorbic acid.
| 1.46 |
| 6 |
| 8.27 |
0.90 |
5.60 |
1.02 |
0.44 |
1.63 |
| 7 |
8.00 |
0.71 |
5.24 |
0.88 |
0.38 |
1.53 |
| 8 |
8.14 |
1.17 |
5.87 |
1.22 |
0.53 |
1.71 |
| 9 |
8.35 |
1.16 |
5.97 |
1.21 |
0.53 |
1.74 |
| 10 |
7.83 |
0.80 |
5.24 |
0.93 |
0.40 |
1.53 |
| DDS |
7.78 |
−1.10 |
3.48 |
0.14 |
0.06 |
1.01 |
| BHT |
7.63 |
−1.64 |
3.04 |
0.05 |
0.02 |
0.89 |
| AA |
9.06 |
−1.16 |
4.14 |
0.19 |
0.08 |
1.21 |
| F |
21.26 |
1.84 |
13.86 |
2.31 |
1.00 |
4.04 |
| Na |
5.38 |
0.40 |
3.43 |
0.54 |
0.23 |
1.00 |
| Compound |
Substituent |
%Reduction |
Ratio Derivative/DDS |
Ratio Derivative/AA |
| 1 |
Hydrogen |
15.0 ± 0.7 ** |
0.9 |
0.15 |
| 2 |
4-hydroxyl |
16.9 ± 1.3 ns |
1.0 |
0.17 |
| 3 |
4-methoxy |
28.4 ± 0.8 *** |
1.6 |
0.28 |
| 4 |
4-nitro |
40.7 ± 0.2 *** |
2.4 |
0.41 |
| 5 |
4-fluoro |
13.2 ± 0.3 *** |
0.8 |
0.13 |
| 6 |
4-chloro |
14.5 ± 0.4 *** |
0.8 |
0.15 |
| 7 |
4-bromo |
19.3 ± 1.4 ** |
1.1 |
0.19 |
| 8 |
4-cyano |
20.0 ± 0.8 *** |
1.2 |
0.20 |
| 9 |
4-carboxyl |
44.8 ± 0.7 *** |
2.6 |
0.45 |
| 10 |
2-phenylethylen |
39.6 ± 0.1 *** |
2.3 |
0.40 |
| DDS |
|
17.3 ± 0.6 |
1.0 |
0.17 |
| AA |
|
100.0 ± 0.3 |
5.8 |
1.00 |