Bile acids are cholesterol-derived metabolites with a well-established role in the digestion and absorption of dietary fat. More recently, the discovery of bile acids as natural ligands for the nuclear farnesoid X receptor (FXR) and membrane Takeda G-protein-coupled receptor 5 (TGR5), and the recognition of the effects of FXR and TGR5 signaling have led to a paradigm shift in knowledge regarding bile acid physiology and metabolic health. Bile acids are now recognized as signaling molecules that orchestrate blood glucose, lipid and energy metabolism. Changes in FXR and/or TGR5 signaling modulates the secretion of gastrointestinal hormones including glucagon-like peptide-1 (GLP-1) and peptide YY (PYY), hepatic gluconeogenesis, glycogen synthesis, energy expenditure, and the composition of the gut microbiome.
- bile acids
- TGR-5
- FXR
- gastrointestinal hormones
- energy intake
- body weight
1. Introduction
Bile acids are synthesized in the liver, where cholesterol is converted via 7α-hydroxylase (CYP7A1) and, to a lesser extent, 27α-hydroxylase (CYP27A1) and 24-hydroxylase (CYP46A1), to the primary bile acids cholic acid (CA) and chenodeoxycholic acid (CDCA) in humans (CA and muricholic acid in rodents). These are then conjugated to glycine or taurine, prior to their secretion into bile [1]. Following meal ingestion, bile acids are released into the gut upon gallbladder emptying, and about 95% of intestinal bile acids is absorbed in the ileum via the apical sodium bile acid co-transporter (ASBT), returning to the liver for re-secretion—a highly efficient process known as “enterohepatic circulation”. A small fraction of bile acids reach the large intestine, where they are modified (through de-conjugation and dihydroxylation) by gut bacteria to secondary bile acids such as deoxycholic acid (DCA), lithocholic acid (LCA), and ursodeoxycholic acid (UDCA, a secondary bile acid in humans, but a primary bile acid in rodents), and absorbed passively into the circulation or excreted in the feces [2] (Figure 1). Bile acids lost to the large intestine are replenished by de novo hepatic synthesis, which is regulated by fibroblast growth factor-19 (FGF19) signaling in the small intestine in humans (or FGF15 in rodents). Thus, bile acids are found in high concentrations in the liver [3], bile [4], and small intestine [5].
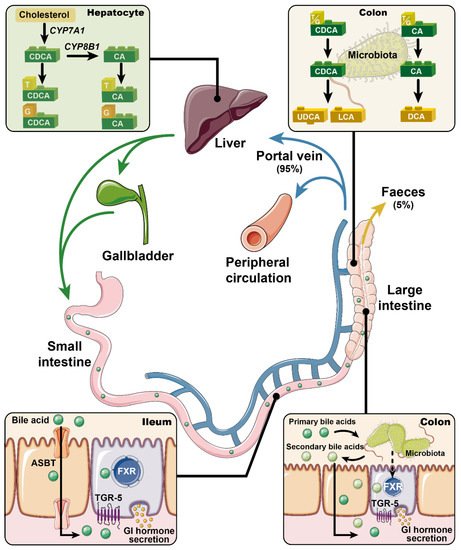
Figure 1. Primary bile acids (i.e., chenodeoxycholic acid (CDCA) and cholic acid (CA)) are synthesized from cholesterol in the liver, and conjugated to glycine and taurine prior to their secretion into bile. In response to meals, bile acids are discharged into the intestine. Approximately 95% of the intestinal bile acids are absorbed in the ileum via apical sodium bile acid co-transporter (ASBT) and return to the liver for re-secretion (i.e., the enterohepatic circulation). Only ~5% of bile acids escape into the large intestine and are modified by gut microbiota into secondary bile acids (e.g., deoxycholic acid (DCA), lithocholic acid (LCA), and ursodeoxycholic acid (UDCA)). Bile acids are now recognized as pivotal signaling molecules that participate in the regulation of metabolic homeostasis through regulating the secretion of gastrointestinal hormones. This complex process has been linked to activation of the nuclear farnesoid X receptor (FXR) and/or the membrane Takeda G-protein-coupled receptor 5 (TGR5). Accordingly, modulation of FXR and/or TGR5 signaling has been actively pursued for the management of metabolic disorders.
For more than a century, bile acids have been regarded solely as “intestinal detergents” that emulsify dietary fat for digestion and transport. The recognition that bile acids are also pivotal signaling molecules orchestrating glucose, lipid and energy metabolism is recent. Bile acids also bind to numerous nuclear and cytoplasmic receptors such as the vitamin D receptor [6], pregnane X receptor [7], and constitutive androstane receptor [8]. However, it was the identification of the bile acid-specific nuclear farnesoid X receptor (FXR) in 1999 and membrane Takeda G-protein-coupled receptor 5 (TGR5) in 2002 that provided a mechanistic framework for a role of BA signaling in the context of metabolism [9][10]. FXR and TGR5 are present in numerous tissues including the central and peripheral nervous systems; bile acid signaling in the latter has been shown to regulate energy intake [11], as supported by the observation that suppression of energy intake induced by intravenous injection of DCA is attenuated when TGR5 was silenced in the vagal nodose ganglia in rats [12]. However, the clinical relevance of this concept is unclear, particularly given that plasma bile acid concentrations are low and that in obese individuals, relative elevation in plasma bile acid levels are not associated with reduced energy intake. In line with the high turnover of bile acids in the enterohepatic circulation, both FXR and TGR5 are expressed abundantly in the liver and the intestine. Signaling through both receptors has been linked to the secretion of gastrointestinal hormones, known to be integral to the maintenance of metabolic homeostasis (Figure 1). For example, the release of ghrelin from gastric G-cells during fasting appears pivotal to sensations of hunger, and stimulation of energy intake. After meals, the secretion of cholecystokinin (CCK) from enteroendocrine I-cells located in the upper gut, and glucagon-like peptide-1 (GLP-1) and peptide YY (PYY) from L-cells located most abundantly in the distal gut, form an integrated signaling system that slows gastrointestinal motility and transit, drives the secretion of insulin to regulate postprandial glucose metabolism (via GLP-1) and suppresses appetite and energy intake [13]. The role of bile acids in the control of blood glucose and lipid metabolism has been reviewed in detail [14][15][16][17], but their potential to impact on the regulation of energy intake has received less attention, despite the recognition, since 1968, that oral administration of CDCA and DCA stimulated PYY secretion and suppressed appetite in obese individuals [18].
2. Effects of Bile Acids on Gastrointestinal Hormone Secretion
The last two decades have witnessed a substantial effort to increase the understanding of the effects of bile acids on gastrointestinal hormone secretion and the consequent impact on metabolism. In healthy individuals, postprandial plasma bile acid concentrations have been reported to correlate negatively with ghrelin, and positively with GLP-1 and PYY [19]. Similar relationships have also been observed in obese patients following bariatric surgery [20]. However, bile acids per se do not appear to affect ghrelin secretion in rats; intestinal infusion of a mixture of physiological bile acids did not affect portal ghrelin levels [21]. In contrast, small intestinal sensing of bile acids has been reported to inhibit CCK secretion in both rodents and humans [22][23], supporting the existence of a negative feedback loop between the two. In contrast, the effects on GLP-1 and PYY release from L-cells have been studied extensively in preclinical and clinical models [24][25][26], stimulating the potential development of bile acid-based interventions for metabolic disorders. While bile acid-induced release of GLP-1 and PYY has been linked to signaling via FXR and TGR5, the data are inconsistent, which may relate to differences in the binding affinity of individual bile acids at FXR and TGR5 (Table 1) and/or complex interactions between the two signaling pathways.
Table 1. Binding affinities of bile acids to human TGR5 and FXR.
| Bile Acid | TGR5 | FXR | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Subjects | Indicator | EC | 50 | Subjects | Indicator | EC | 50 | ||||||
| Primary Bile Acids | |||||||||||||
| CA | CHO cells/HEK293 | Intracellular cAMP | 7.72 µM | [27] | / >10 µM | [10] | CV-1 cells | Reporter gene activation | No effect | [28] | |||
| CDCA | CHO cells/HEK293 | Intracellular cAMP | 4.43 µM | [27] | / 4 µM | [10] | HepG2 cells /CV-1 cells |
Reporter gene activation | 10 µM | [9] | / 50 µM | [28] | |
| CHO cells | Reporter gene activation | 6.71 µM | [29] | Cell-free | Ligand-sensing assay | 4.5 µM | [30] | ||||||
| Conjugated Primary Bile Acids | |||||||||||||
| TCA/GCA | CHO cells | Reporter gene activation | 4.95 µM/ 13.6 µM | [29] | Cell-free | Ligand-sensing assay | No effect | [30] | |||||
| TCDCA/ | GCDCA | CHO cells | Reporter gene activation | 1.92 µM/ 3.88 µM | [29] | Cell-free | Ligand-sensing assay | 10 µM | [30] | ||||
| HCA | Cell-free | TR-FRET FXR coactivator assay | 70.06 µM (IC | 50 | ) | [31] | |||||||
| Secondary bile acids | |||||||||||||
| DCA | CHO cells | Intracellular cAMP | 1.01 µM | [27] | HepG2 cells | Reporter gene activation | 100 µM | [9] | |||||
| HEK293 | Intracellular cAMP | 575 nM | [10] | CV-1 cells | Reporter gene activation | 50 µM | [28] | ||||||
| LCA | CHO cells | Intracellular cAMP | 0.53 µM | [27] | CV-1 cells | Reporter gene activation | 50 µM | [28] | |||||
| HEK293 | Intracellular cAMP | 35 nM | [10] | Cell-free | Ligand-sensing assay | 25 µM | [6] | ||||||
| UDCA | CHO cells | Reporter gene activation/Intracellular cAMP | 36.4 µM | [29] | / No effect | [27] | CV-1 cells | Reporter gene activation | No effect | [28] | |||
| HDCA | CHO cells | Reporter gene activation | 31.6 µM | [29] | Cell-free | TR-FRET FXR coactivator assay | 62.43 µM | [31] | (IC | 50 | ) | ||
| Conjugated Secondary Bile Acids | |||||||||||||
| TDCA/ | GDCA | CHO cells | Reporter gene activation | 0.79 µM /1.18 µM | [29] | Cell-free | Ligand-sensing assay | 500 µM | [30] | (IC | 50 | ) | |
| TLCA/ | GLCA | CHO cells | Reporter gene activation | 0.29 µM /0.54 µM | [29] | Cell-free | Ligand-sensing assay | 3.8 µM/4.7 µM | [30] | (IC | 50 | ) | |
| TUDCA/ | GUDCA | CHO cells | Reporter gene activation | 30.0 µM /33.9 µM | [29] | Cell-free | Ligand-sensing assay | No effect | [30] | ||||
| THDCA/GHDCA | CHO cells | Reporter gene activation | 24.2 µM/36.7 µM | [29] | |||||||||
Note: EC50: the concentration for a half maximal effect; IC50: the concentration for a half maximal inhibitory effect; CHO: Chinese hamster ovary cells; HepG2 cells: Human hepatoma cell line; CV-1 cells: Monkey kidney fibroblast cells (CV-1 line); HEK293: human embryonic kidney cell line 293; TR-FRET FXR coactivator assay: commercial assay kit for screening ligand for FXR.
2.1. FXR
FXR is expressed abundantly in the liver and the intestine, and the binding affinity of individual bile acids is variable (CDCA > DCA > LCA > CA > UDCA, Table 1). FXR was initially identified as a regulator of bile acid metabolism [14], and subsequently as a modulator of L-cell secretion. Indeed, FXR is expressed by the murine L-cell line, GLUTag. However, the FXR agonist GW4064 and CDCA (which preferentially binds FXR) were shown to suppress glucose-induced proglucagon expression and GLP-1 secretion in this cell line by decreasing glycolysis, whereas silencing FXR abolished these effects [32]. These observations have been replicated in studies with different L-cell lines (i.e., NCI-H736 [31] and STC-1 [33]). In a similar manner, GW4064 blunted the GLP-1 response to short-chain fatty acids (SCFA) in both GLUTag and NCI-H716 cell lines [34]. Consistent with these observations, FXR-deficient mice exhibited increased GLP-1 secretion in response to both dietary fiber, which increases colonic SCFA [34], and oral glucose [35]. Oral intake of GW4064 (10 mg/kg, 2 doses over 12 h) also decreased active GLP-1 levels in the plasma of mini-pigs [31]. However, in an isolated perfusion model of rat intestine, both luminal and vascular perfusion of GW4064 failed to affect the GLP-1 response to a physiological mixture of bile acids in rats [21]. In mice, diversion of bile acids from the gallbladder to the ileum was shown to modestly increase GLP-1 secretion, improve glucose tolerance, and induce weight loss [36]. The reductions in postprandial blood glucose and body weight induced by this procedure were abolished in intestinal FXR-knockout mice, suggesting that intestinal FXR-signaling can potentially promote GLP-1 secretion. Unfortunately, the study failed to determine whether the rise in GLP-1 was specifically induced by FXR-activation [36]. Of note, oral administration of the intestine-restricted FXR agonist, fexaramine, in mice was reported to increase the abundance of LCA-producing gut bacteria to activate TGR5-signaling indirectly, leading to enhanced GLP-1 secretion and improvement in insulin sensitivity and lipid profile as well as the promotion of adipose tissue browning [37]. Accordingly, outcomes derived from ex vivo and in vivo experiments are, by and large, inconsistent, although the intestine-restricted FXR signaling appears to have an overall favorable effect on metabolic health.
2.2. TGR5
TGR5, also known as GPBAR1, is a G-protein coupled receptor that is expressed widely in the gastrointestinal tract, pancreas, liver, gallbladder, and adipose tissue. Like FXR, its binding affinity for individual bile acids varies substantially (LCA > DCA > CDCA > CA > UDCA, Table 1) [27]. TGR5 activation has been reported to suppress hepatic macrophages, induce gallbladder relaxation and refilling, and promote intestinal motility [14]. TGR5 is also expressed on L-cells. Unlike FXR, stimulation of TGR5 by LCA and DCA was shown to potently stimulate GLP-1 secretion from STC-1 cells in a dose-dependent manner, an effect suppressed by downregulation of TGR5 expression [38]. The stimulatory effect of TGR5 on GLP-1 secretion required the closure of ATP-sensitive potassium (KATP) channels and elevated intracellular concentrations of cAMP and Ca2+ [39][40]. A major observation in relation to TGR5 signaling was the demonstration of its basolateral location on L-cells. Thus, to activate TGR5, it is necessary for bile acids or other TGR5 ligands to be transported through the epithelial layer [41]. However, the readily absorbed TGR5 agonist SB-756050 failed to stimulate GLP-1 secretion significantly, or improve glycemic control at various doses compared with the placebo in acute studies involving patients with T2D [42]. It is noteworthy that L-cells are distributed most densely in the distal gut regions [13]. It would therefore be of interest to investigate whether delivery of TGR5 agonists should be targeted at the distal gut.
PYY is co-released with GLP-1 from L-cells, and it was initially noted that perfusion of DCA (1–25 mM) into the isolated rabbit colon increased PYY secretion substantially in a dose-dependent manner [18]. Intracolonic administration of DCA or TCA in humans has also been shown to induce a rapid and substantial rise in plasma PYY [43][44][45]. Similar to TGR5-mediated GLP-1 secretion, the outcomes of studies using isolated rat colon indicate that bile acid-induced PYY secretion is dependent on bile acid translocation from the luminal to basolateral side [46]. That PYY secretion is less evident in response to bile acids with poor affinity to TGR5, and attenuated in TGR5-knockout models, attests to the fundamental relevance of TGR5-signaling to bile acid-induced PYY secretion [47].
In summary, there is compelling evidence for a role of bile acids in the modulation of GLP-1 and PYY secretion in both animals and humans. Stimulation of TGR5 on L-cells induces the secretion of both hormones, while effects of FXR signaling remain controversial. The interactions between FXR and TGR5 signaling remain poorly characterized and an improved understanding may be of relevance to the development of novel strategies for the management of metabolic disorders.
References
- Russell, D.W. The enzymes, regulation, and genetics of bile acid synthesis. Annu. Rev. Biochem. 2003, 72, 137–174.
- Thomas, C.; Pellicciari, R.; Pruzanski, M.; Auwerx, J.; Schoonjans, K. Targeting bile-acid signalling for metabolic diseases. Nat. Rev. Drug Discov. 2008, 7, 678–693.
- Setchell, K.D.; Rodrigues, C.M.; Clerici, C.; Solinas, A.; Morelli, A.; Gartung, C.; Boyer, J. Bile acid concentrations in human and rat liver tissue and in hepatocyte nuclei. Gastroenterology 1997, 112, 226–235.
- Shiffman, M.L.; Sugerman, H.J.; Kellum, J.M.; Moore, E.W. Changes in gallbladder bile composition following gallstone formation and weight reduction. Gastroenterology 1992, 103, 214–221.
- Northfield, T.C.; McColl, I. Postprandial concentrations of free and conjugated bile acids down the length of the normal human small intestine. Gut 1973, 14, 513–518.
- Makishima, M.; Lu, T.T.; Xie, W.; Whitfield, G.K.; Domoto, H.; Evans, R.M.; Haussler, M.R.; Mangelsdorf, D.J. Vitamin D receptor as an intestinal bile acid sensor. Science 2002, 296, 1313–1316.
- Ihunnah, C.A.; Jiang, M.; Xie, W. Nuclear receptor PXR, transcriptional circuits and metabolic relevance. Biochim. Biophys. Acta 2011, 1812, 956–963.
- Wagner, M.; Halilbasic, E.; Marschall, H.U.; Zollner, G.; Fickert, P.; Langner, C.; Zatloukal, K.; Denk, H.; Trauner, M. CAR and PXR agonists stimulate hepatic bile acid and bilirubin detoxification and elimination pathways in mice. Hepatology 2005, 42, 420–430.
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a nuclear receptor for bile acids. Science 1999, 284, 1362–1365.
- Maruyama, T.; Miyamoto, Y.; Nakamura, T.; Tamai, Y.; Okada, H.; Sugiyama, E.; Nakamura, T.; Itadani, H.; Tanaka, K. Identification of membrane-type receptor for bile acids (M-BAR). Biochem. Biophys. Res. Commun. 2002, 298, 714–719.
- Mertens, K.L.; Kalsbeek, A.; Soeters, M.R.; Eggink, H.M. Bile acid signaling pathways from the enterohepatic circulation to the central nervous system. Front. Neurosci. 2017, 11, 617.
- Wu, X.; Li, J.-Y.; Lee, A.; Lu, Y.-X.; Zhou, S.-Y.; Owyang, C. Satiety induced by bile acids is mediated via vagal afferent pathways. JCI Insight 2020, 5.
- Xie, C.; Jones, K.L.; Rayner, C.K.; Wu, T. Enteroendocrine hormone secretion and metabolic control: Importance of the region of the gut stimulation. Pharmaceutics 2020, 12, 790.
- Lefebvre, P.; Cariou, B.; Lien, F.; Kuipers, F.; Staels, B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol. Rev. 2009, 89, 147–191.
- Fiorucci, S.; Mencarelli, A.; Palladino, G.; Cipriani, S. Bile-acid-activated receptors: Targeting TGR5 and farnesoid-X-receptor in lipid and glucose disorders. Trends Pharmacol. Sci. 2009, 30, 570–580.
- Kuipers, F.; Bloks, V.W.; Groen, A.K. Beyond intestinal soap—Bile acids in metabolic control. Nat. Rev. Endocrinol. 2014, 10, 488–498.
- Ahmad, T.R.; Haeusler, R.A. Bile acids in glucose metabolism and insulin signalling–mechanisms and research needs. Nat. Rev. Endocrinol. 2019.
- Bray, G.A.; Gallagher, T.F., Jr. Suppression of appetite by bile acids. Lancet 1968, 1, 1066–1067.
- Roberts, R.E.; Glicksman, C.; Alaghband-Zadeh, J.; Sherwood, R.A.; Akuji, N.; le Roux, C.W. The relationship between postprandial bile acid concentration, GLP-1, PYY and ghrelin. Clin. Endocrinol. 2011, 74, 67–72.
- Nakatani, H.; Kasama, K.; Oshiro, T.; Watanabe, M.; Hirose, H.; Itoh, H. Serum bile acid along with plasma incretins and serum high-molecular weight adiponectin levels are increased after bariatric surgery. Metabolism 2009, 58, 1400–1407.
- Kuhre, R.E.; Wewer Albrechtsen, N.J.; Larsen, O.; Jepsen, S.L.; Balk-Moller, E.; Andersen, D.B.; Deacon, C.F.; Schoonjans, K.; Reimann, F.; Gribble, F.M.; et al. Bile acids are important direct and indirect regulators of the secretion of appetite- and metabolism-regulating hormones from the gut and pancreas. Mol. Metab. 2018, 11, 84–95.
- Gomez, G.; Upp, J.R., Jr.; Lluis, F.; Alexander, R.W.; Poston, G.J.; Greeley, G.H., Jr.; Thompson, J.C. Regulation of the release of cholecystokinin by bile salts in dogs and humans. Gastroenterology 1988, 94, 1036–1046.
- Koop, I.; Schindler, M.; Bosshammer, A.; Scheibner, J.; Stange, E.; Koop, H. Physiological control of cholecystokinin release and pancreatic enzyme secretion by intraduodenal bile acids. Gut 1996, 39, 661–667.
- Sonne, D.P.; Hansen, M.; Knop, F.K. Bile acid sequestrants in type 2 diabetes: Potential effects on GLP1 secretion. Eur. J. Endocrinol. 2014, 171, R47–R65.
- Guida, C.; Ramracheya, R. PYY, a therapeutic option for type 2 diabetes? Clin. Med. Insights Endocrinol. Diabetes 2020, 13, 1179551419892985.
- Wu, T.; Bound, M.J.; Standfield, S.D.; Jones, K.L.; Horowitz, M.; Rayner, C.K. Effects of taurocholic acid on glycemic, glucagon-like peptide-1, and insulin responses to small intestinal glucose infusion in healthy humans. J. Clin. Endocrinol. Metab. 2013, 98, E718–E722.
- Kawamata, Y.; Fujii, R.; Hosoya, M.; Harada, M.; Yoshida, H.; Miwa, M.; Fukusumi, S.; Habata, Y.; Itoh, T.; Shintani, Y.; et al. A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 2003, 278, 9435–9440.
- Wang, H.; Chen, J.; Hollister, K.; Sowers, L.C.; Forman, B.M. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol. Cell 1999, 3, 543–553.
- Sato, H.; Macchiarulo, A.; Thomas, C.; Gioiello, A.; Une, M.; Hofmann, A.F.; Saladin, R.; Schoonjans, K.; Pellicciari, R.; Auwerx, J. Novel potent and selective bile acid derivatives as TGR5 agonists: Biological screening, structure-activity relationships, and molecular modeling studies. J. Med. Chem. 2008, 51, 1831–1841.
- Parks, D.J.; Blanchard, S.G.; Bledsoe, R.K.; Chandra, G.; Consler, T.G.; Kliewer, S.A.; Stimmel, J.B.; Willson, T.M.; Zavacki, A.M.; Moore, D.D.; et al. Bile acids: Natural ligands for an orphan nuclear receptor. Science 1999, 284, 1365–1368.
- Zheng, X.; Chen, T.; Jiang, R.; Zhao, A.; Wu, Q.; Kuang, J.; Sun, D.; Ren, Z.; Li, M.; Zhao, M.; et al. Hyocholic acid species improve glucose homeostasis through a distinct TGR5 and FXR signaling mechanism. Cell Metab. 2020.
- Trabelsi, M.S.; Daoudi, M.; Prawitt, J.; Ducastel, S.; Touche, V.; Sayin, S.I.; Perino, A.; Brighton, C.A.; Sebti, Y.; Kluza, J.; et al. Farnesoid X receptor inhibits glucagon-like peptide-1 production by enteroendocrine L cells. Nat. Commun. 2015, 6, 7629.
- Li, P.; Zhu, L.; Yang, X.; Li, W.; Sun, X.; Yi, B.; Zhu, S. Farnesoid X receptor interacts with cAMP response element binding protein to modulate glucagon-like peptide-1 (7-36) amide secretion by intestinal L cell. J. Cell Physiol. 2019, 234, 12839–12846.
- Ducastel, S.; Touche, V.; Trabelsi, M.S.; Boulinguiez, A.; Butruille, L.; Nawrot, M.; Peschard, S.; Chavez-Talavera, O.; Dorchies, E.; Vallez, E.; et al. The nuclear receptor FXR inhibits Glucagon-Like Peptide-1 secretion in response to microbiota-derived Short-Chain Fatty Acids. Sci. Rep. 2020, 10, 174.
- Xie, C.; Jiang, C.; Shi, J.; Gao, X.; Sun, D.; Sun, L.; Wang, T.; Takahashi, S.; Anitha, M.; Krausz, K.W.; et al. An intestinal farnesoid X receptor-ceramide signaling axis modulates hepatic gluconeogenesis in mice. Diabetes 2017, 66, 613–626.
- Albaugh, V.L.; Banan, B.; Antoun, J.; Xiong, Y.; Guo, Y.; Ping, J.; Alikhan, M.; Clements, B.A.; Abumrad, N.N.; Flynn, C.R. Role of bile acids and GLP-1 in mediating the metabolic improvements of bariatric surgery. Gastroenterology 2019, 156, 1041–1051.e1044.
- Pathak, P.; Xie, C.; Nichols, R.G.; Ferrell, J.M.; Boehme, S.; Krausz, K.W.; Patterson, A.D.; Gonzalez, F.J.; Chiang, J.Y.L. Intestine farnesoid X receptor agonist and the gut microbiota activate G-protein bile acid receptor-1 signaling to improve metabolism. Hepatology 2018, 68, 1574–1588.
- Katsuma, S.; Hirasawa, A.; Tsujimoto, G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem. Biophys. Res. Commun. 2005, 329, 386–390.
- Thomas, C.; Gioiello, A.; Noriega, L.; Strehle, A.; Oury, J.; Rizzo, G.; Macchiarulo, A.; Yamamoto, H.; Mataki, C.; Pruzanski, M.; et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009, 10, 167–177.
- Parker, H.E.; Wallis, K.; le Roux, C.W.; Wong, K.Y.; Reimann, F.; Gribble, F.M. Molecular mechanisms underlying bile acid-stimulated glucagon-like peptide-1 secretion. Br. J. Pharmacol. 2012, 165, 414–423.
- Brighton, C.A.; Rievaj, J.; Kuhre, R.E.; Glass, L.L.; Schoonjans, K.; Holst, J.J.; Gribble, F.M.; Reimann, F. Bile acids trigger GLP-1 release predominantly by accessing basolaterally located G protein-coupled bile acid receptors. Endocrinology 2015, 156, 3961–3970.
- Hodge, R.J.; Lin, J.; Vasist Johnson, L.S.; Gould, E.P.; Bowers, G.D.; Nunez, D.J.; Team, S.B.P. Safety, pharmacokinetics, and pharmacodynamic effects of a selective TGR5 agonist, SB-756050, in type 2 diabetes. Clin. Pharmacol. Drug Dev. 2013, 2, 213–222.
- Adrian, T.; Ballantyne, G.; Longo, W.; Bilchik, A.; Graham, S.; Basson, M.; Tierney, R.; Modlin, I. Deoxycholate is an important releaser of peptide YY and enteroglucagon from the human colon. Gut 1993, 34, 1219–1224.
- Adrian, T.E.; Gariballa, S.; Parekh, K.A.; Thomas, S.A.; Saadi, H.; Al Kaabi, J.; Nagelkerke, N.; Gedulin, B.; Young, A.A. Rectal taurocholate increases L cell and insulin secretion, and decreases blood glucose and food intake in obese type 2 diabetic volunteers. Diabetologia 2012, 55, 2343–2347.
- Wu, T.; Bound, M.J.; Standfield, S.D.; Gedulin, B.; Jones, K.L.; Horowitz, M.; Rayner, C.K. Effects of rectal administration of taurocholic acid on glucagon-like peptide-1 and peptide YY secretion in healthy humans. Diabetes Obes. Metab. 2013, 15, 474–477.
- Tough, I.R.; Schwartz, T.W.; Cox, H.M. Synthetic G protein-coupled bile acid receptor agonists and bile acids act via basolateral receptors in ileal and colonic mucosa. Neurogastroenterol. Motil. 2020, 32, e13943.
- Christiansen, C.B.; Trammell, S.A.J.; Wewer Albrechtsen, N.J.; Schoonjans, K.; Albrechtsen, R.; Gillum, M.P.; Kuhre, R.E.; Holst, J.J. Bile acids drive colonic secretion of glucagon-like-peptide 1 and peptide-YY in rodents. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G574–G584.