Feline calicivirus (FCV) is an important pathogen of cats that has two genogroups (GI and GII). To investigate the prevalence and molecular characteristics of FCVs in southwestern China, 162 nasal swab samples were collected from cats in animal shelters and pet hospitals. In total, 38 of the clinical samples (23.46%) were identified as FCV positive using nested RT-PCR. Phylogenetic analyses using 10 capsid protein VP1 sequences revealed that 8 GI and 2 GII strains formed two independent clusters. Additionally, three separated FCVs that not cluster phylogenetically (two GI and one GII strains) were successfully isolated from clinical samples and their full-length genomes were obtained. Phylogenetic and recombinant analyses of a GI FCV revealed genomic breakpoints in ORF1 and ORF2 regions with evidence for recombinant events between GI sub-genogroups, which is reported in China for the first time. Furthermore, sera obtained from mice immunized independently with the three FCV isolates and a commercial vaccine were used to evaluate the cross-reactivity of neutralizing antibodies. The three separate FCVs were neutralized by each other at a 1:19 to 1:775 titer rang, whereas the triple-inactivated vaccine was at a titer of 1:16, which suggested that different genogroup/sub-genogroup FCV strains exhibit significantly different titers of neutralizing antibodies, including the commercial FCV vaccine. Thus, our study revealed the genetic diversity and complex cross-reactivity levels of FCVs in southwestern China, which provides new insights into applying in vaccination strategies.
1. Introduction
Feline calicivirus (FCV), which belongs to the family
Caliciviridae, genus
Vesivirus, is the causative agent of respiratory disease, stomatitis, arthritis, and hemorrhagic-like fever in cats
[1]. The virus was first isolated in 1957
[2] and is now widely distributed in many European, American, and Asian counties
[3,4,5][3][4][5]. Generally, an FCV infection causes apparent or relatively mild oral cavity and upper respiratory tract symptoms
[6,7][6][7]. Occasionally, FCV causes ulcerative dermatitis, severe pneumonia, feline chronic gingivo-stomatitis complex, abortion, acute jaundice, and acute febrile lameness syndrome
[8,9][8][9]. However, in the past decade, virulent mutants of FCV called VS-FCVs, have been identified as the causal agents of severe and acute virulent systemic disease (VSD), resulting in a high persistent fever and high mortality rate in vaccinated cats
[10,11][10][11].
The FCV genome is a non-enveloped, single-stranded positive-sense RNA of approximately 7.7 kb having three open reading frames (ORFs), including ORF1, ORF2, and ORF3
[12]. The ORF2 (2013 nt) encodes the capsid protein VP1 and is divided into six regions (A–F). The regions A, B, D, and F are relatively well conserved, whereas regions C and E are variable with significant sequence divergence levels
[13]. Therefore, the
VP1 gene is frequently used as a target in molecular epidemiological investigations
[4,14,15][4][14][15]. In accordance with the genetic diversity of the
VP1 gene, the global FCVs are classified into two genogroups: genogroup I (GI) and genogroup II (GII)
[14]. In China, most FCV strains belong to GII, and they are closely related to Japanese FCV strains
[5,16][5][16]. It is worthy to note that new FCV strains have been constantly emerging in recent years owing to the large degree of viral genetic variation
[17,18,19][17][18][19].
Vaccination against FCV has been the mainstay control method for decades
[20,21,22][20][21][22]. Although several commercial vaccines are widely used in the field and effectively reduce the clinical disease incidence, it is not clear whether the existing commercial vaccines have cross-protection against the newly emerging FCV strains.
2. RT-PCR Survey of Clinical Samples
In this study, 38 out of the 162 clinical samples were identified as positive for FCV using nested RT-PCR amplification, resulting in a positivity rate of 23.46% (38/162, 95% CI: 17.2–30.7%). Among them, 30 samples tested positive from 84 cats in 14 animal shelters (positive rate: 35.71%, 95% CI: 25.6–46.9%), and 8 samples tested positive from 78 cats in 2 pet hospitals (positive rate: 10.26%, 95% CI: 4.5–19.2%)
(Table 1). Thus, the cats in the animal shelters had a significantly higher positive rate than those in the pet hospitals (
p < 0.05).
3. Sequence Alignment and Phylogenetic Analysis of the VP1 Gene
Of the 38 positive samples, 10 VP1 hypervariable regions (HVs) were successfully obtained by sequencing and designated as SMU-B5, SMU-Y5, SMU-B9, SMU-B22, SMU-F4, SMU-Y14, SMU-C17, SMU-20, SMU-R1, and SMU-R55 (GenBank no.: MW194992–MW195001). Sequence alignment showed that the nucleotide similarity of the 10 VP1 HVs ranged from 72.5% to 92.8%, with 80.9–96.8% similarity levels in the deduced amino acid sequences. In addition, these viruses shared 72.0–89.2% nucleotide homology and 77.8–92.4% amino acid homology with the 35 reference FCV sequences from GenBank.
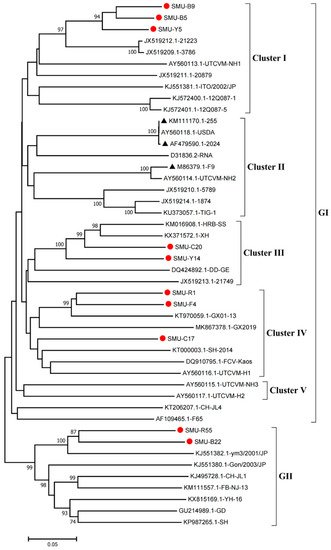
Phylogenetic analysis showed that the FCV strains clustered into two genogroups, GI and GII, as previously described
[14], and the GI group is further classified into five clusters (I-V). Eight strains (SMU-B5, SMU-Y5, SMU-B9, SMU-Y14, SMU-C20, SMU-C17, SMU-R1, and SMU-F4) were divided into three clusters in the GI group. Among these strains, SMU-B5, SMU-Y5, and SMU-B9 are closely related to strain FCV-21223 from the USA, and SMU-Y14 and SMU-C20 share close genetic relationships with the strains HRB-SS and XH from China. Additionally, three strains (SMU-R1, SMU-F4, and SMU-C17) were classified into a minor branch together with the Chinese strains GX2019 and GX01–13 from Guangxi province. The remaining two strains (SMU-R55 and SMU-B22) belong to the GII group and are closely related to strain ym3/2001/JP from Japan (
Figure 1). No strains in this study were divided into cluster II of the GI group, which contains the representative vaccine strains FCV-255 (KM111170), FCV-2024 (AF479590), and F9 (M86379), suggesting that the FCV strains identified in vaccinated cats certainly cannot be related to used vaccines. Thus, FCV strains present in southwestern China are genetically diverse and complex.
Figure 1. Phylogenetic tree of FCVs from southwestern China and 35 reference strains based on nucleotide sequences of VP1 HV. The phylogenetic tree was constructed with MEGA7.1 software (Tempe, AZ, USA) by the neighbor-joining method with 1000 bootstrap values. The viral assembly sequences characterized in this study are marked by “red circles” and the vaccine strains are labeled with “black triangles”. The scale bar indicates the number of nucleotide substitutions per site.
4. Isolation and Identification of FCV Strains
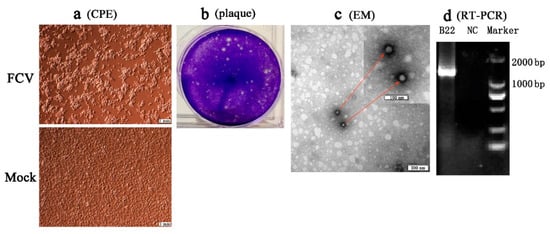
Four separated strains (SMU-B22, SMU-F4, SMU-Y14, and SMU-B5) that did not cluster phylogenetically were filtered out for further viral isolation, and three strains (designated SMU-B22-2020, SMU-F4-2020, and SMU-B5-2020) were successfully isolated from the clinical samples using CRFK cells. The infected CRFK cells exhibited cytogamy, cell rounding, karyopyknosis, and cluster-like forms; they eventually detached completely and remained suspended (
Figure 2a, SMU-B22-2020 isolate shown only). No obvious CPE was observed in the control cells (
Figure 2b). The three isolates were subjected to electron microscope observation after three rounds of plaque purification. Spherical viral particles with no envelopes were observed, and these features are consistent with the structural characteristics of caliciviruses (
Figure 2c). The viruses were further conformed by RT-PCR using the FCV specific primers (
Figure 2d). The TCID
50 values of FCV SMU-B22-2020, SMU-F4-2020, and SMU-B5-2020 were determined as 1 × 10
9.5/0.1, 1 × 10
11/0.1, and 1 × 10
8.5/0.1 mL, respectively.
Figure 2. Isolation and identification of an FCV strain (SMU-B22-2020) in this study. (a) CRFK cells were inoculated with FCV SMU-B22-2020 and uninfected medium is shown at 12 h postinfection; (b) plaque assay for SMU-B22-2020 isolate with a dilution of 10−10 of virus stock using CRFK cells; (c) FCV particles with a diameter of approximately 30 nm were observed in cell supernatants by transmission electron microscopy (EM). Red arrows indicate the FCV particles; (d) the FCV isolate was further confirmed by RT-PCR with a 1300 bp target fragment. NC indicates the negative control.
5. Full-Length Genome Sequence Analyses of Three New FCV Isolates
To further understand the genetic variation of the FCV strains in southwestern China, the full-length or nearly full-length genomes of three novel strains were successfully amplified from the positive FCV samples. The results have been submitted to the GenBank database (accession no.: MW194989–MW194991), and showed 7653 bp, 7686 bp, and 7705 bp in length, respectively. The genetic analyses using MegAlign software revealed that the nucleotide identity ranged from 76.8% to 80.9%, and the sequences shared 76.1–82.9% nucleotide identity levels with 31 other full-length genome reference sequences.
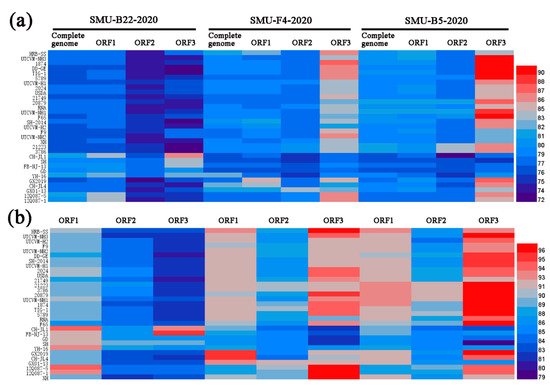
To examine the genomic characteristics of the three FCV strains, the nucleotide (nt) and amino acid (aa) homology levels of the FCV ORFs among the three strains in this study and with the 31 representative strains were compared (
Figure 3a,b). For SMU-B22-2020, ORF1 and ORF3 shared 83.7–86.6% nt (93.5–94.5% aa) homology levels with CH-JL1, which was the highest homology level among the reference strains, whereas ORF2 of SMU-B22-2020 shared a 78.7% nt homology with YH-16 and an 89.2% aa homology with SH, which was a higher percentage than with other reference strains. For SMU-F4-2020, ORF1–3 shared 85.0%/95.5%, 83.5%/91.4%, and 86.3%/97.2% nt/aa homology levels with GX2019, GX01–13, and 12Q087–5, respectively, which were higher than the homology levels with other reference strains. For SMU-B5-2020, ORF1–3 shared their highest nt/aa homology levels with HRB-SS (81.9%/93.2%), FCV-21223 (83.2%/91.8%), and UTCVM-NH3 (91.3%/97.2%), respectively. Thus, recombination events among different viruses might have occurred in their genomes.
Figure 3. A heatmap was constructed based on the nucleotide sequence (a) and amino acid identities (b) of each genome among different FCV strains using HemI software. The colors represent sequence identity (blue for lower identities and red for higher identities) among different strains. The bar on the right represents the hierarchical similarity among the 15 meta-clusters (metric, Euclidean distance; linkage, average).
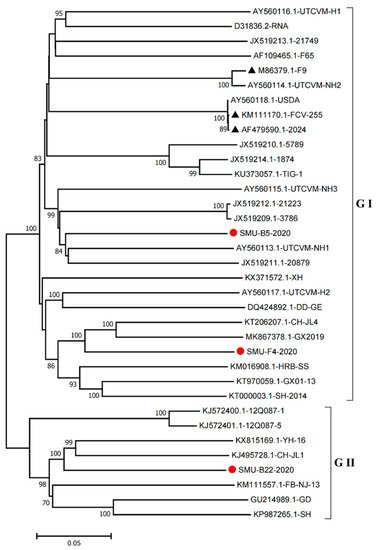
Furthermore, we constructed phylogenetic trees using the complete genomes to determine the genetic relationships of the three isolates. The complete genome of the SMU-B5-2020 strain belonged to cluster III of the GI group and shared a close genetic relationship with two USA strains, UTCVM-NH1 and 20879. The SMU-F4-2020 strain clustered in cluster IV of the GI group and shared close relationships with two Chinese strains, CH-JL4 (northeastern China) and GX2019 (southern China). In addition, the SMU-B22-2020 strain was the neighbor of YH-16 and CH-JL1 in the GII group (
Figure 4). The results suggest that various FCV strains from different clusters are simultaneously circulating and spreading in southwestern China.
Figure 4. Phylogenetic tree based on full-length genomic sequence of three isolates (SMU-B22-2020, SMU-F4-2020, and SMU-B5-2020) and FCV reference strains available in GenBank. The three isolates in this study are labeled with “red circles” and the vaccine strains are labeled with “black triangles”. The scale bar indicates the number of nucleotide substitutions per site.
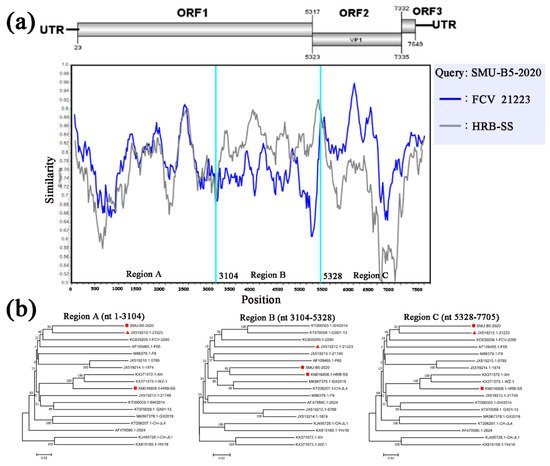
To identify possible recombinant events, we detected recombination using SimPlot and RDP4 software packages. The SMU-B5-2020 strain showed potential recombinant events between the FCV-21223 strain (cluster I of GI) and the HRB-SS strain (cluster III of GI) with
p-values of <1 × 10
−6 from the results of five detection methods. From the similarity plot, two recombination breakpoints within the SMU-B5-2020 genome were identified in ORF1 (nt 3104) and ORF2 (nt 5328) (
Figure 5a). The breakpoints in SMU-B5-2020 separated its genome into three regions, with regions A (nt 1–3104) and C (nt 5328–7705) being closely related to the FCV-21223 strain, whereas region B (nt 3104–5328) was closely related to the HRB-SS strain (
Figure 5b). The results indicated that the SMU-B5-2020 strain likely originated from recombination events between members of clusters I and III of the GI FCV strains.
Figure 5. Full-length genome recombination analysis of SMU-B5-2020. (a) Genome scale similarity comparisons of SMU-B5-2020 (query) with FCV-21223 (blue) and HRB-SS (gray). The supposed recombination region is shown with red colors and the recombination breakpoints are marked at the bottom with nucleotide sites and viral genome structure referenced to FCV-21223; (b) phylogenetic trees based on every recombinant fragment within SMU-B5-2020 and 17 reference FCV strains are shown below the similarity plot. The strain SMU-B5-2020 is labeled with “red circles” and the putative recombinant parent strains are marked with “red triangles”.
6. Serum Neutralization Antibody Titers
Viral antigen was prepared by mixing inactivated FCVs with Freund’s complete adjuvant. A serum virus neutralization assay showed that the SMU-F4-2020-neutralizing antibody titer level in three mice reached as high as 1:775 ± 65 to the SMU-F4-2020 isolate, which was significantly higher than the titers to SMU-B22-2020, SMU-B5-2020, and the vaccine (
p < 0.05). Similarly, the SMU-B22-2020-neutralizing antibody titer level reached 1:655 ± 55 to the SMU-B22-2020 strain, which was significantly higher than the titers to the other two isolates and the commercial vaccine (
p < 0.05). The SMU-B5-2020-neutralizing antibody titer was 1:416 ± 7.5 to SMU-B5-2020, which was significantly higher than the titers to the other two isolates and the commercial vaccine (
p < 0.05) (
Table 21). The results indicated that the FCV-antibodies against each isolate did not provide better cross-reactivity between heterologous FCV isolates.
Table 21.
Determination of serum neutralizing antibody titers to FCV isolates in mice.
| Neutralizing Antibody (NA) |
FCV Isolates |
| SMU-F4-2020 |
SMU-B22-2020 |
SMU-B5-2020 |
| SMU-F4-2020-NA |
1:775 ± 65 | a |
1:21.5 ± 2.5 |
1:19 ± 0 |
| SMU-B22-2020-NA |
1:106.6 ± 0.5 |
1:655 ± 55 |
1:63.5 ± 0.5 |
| SMU-B5-2020-NA |
1:95 ± 5 |
1:54.5 ± 9.5 |
1:416.5 ± 7.5 |
| Vaccine-NA |
1:16 ± 0 |
1:16 ± 0 |
1:16 ± 0 |
a The neutralizing antibody titer is shown as mean ± standard deviation (SD).