Vitamin E was first discovered by the American endocrinologist and anatomist Herbert. M. Evans, together with his assistant Katherine S. Bishop. The isolated substance, later termed vitamin E, describes a group of compounds consisting of four tocopherol (TP)- and four tocotrienol (TT)-derivatives. Considering the consequences of vitamin E deficiency—ataxia, dysarthria and neuromuscular disorders [6]—it is clear that this substance plays an important role in the central and peripheral nervous systems.
- vitamin E
- neurodegenerative diseases
- nutrition
- vitamin E supplementation
- Alzheimer’s disease
- personalized medicine
1. Introduction
Vitamin E was first discovered by the American endocrinologist and anatomist Herbert. M. Evans, together with his assistant Katherine S. Bishop [1]. The isolated substance, later termed vitamin E, describes a group of compounds consisting of four tocopherol (TP)- and four tocotrienol (TT)-derivatives. They all share a chromanol ring as their structural basis and are therefore termed tocochromanols. The chromanol ring is hydroxylated in position 6 and, due to the methylation of the ring, α-, β-, γ- and δ-forms can be differentiated ( Figure 1 ). TPs and TTs are classified according to their side-chain: TPs contain one saturated fatty acid, whereas TTs contain a triple unsaturated fatty acid ( Figure 1 ). The biological properties of vitamin E compounds are mainly determined by their structure. In humans, α-TP is the biologically most active form that binds with highest affinity to the α-tocopherol-transfer protein (α-TTP) [2]. α-TTP is a soluble protein found in the cytosol of hepatocytes in humans that acts to transport TPs between membrane vesicles, allowing for the distribution of vitamin E [3].
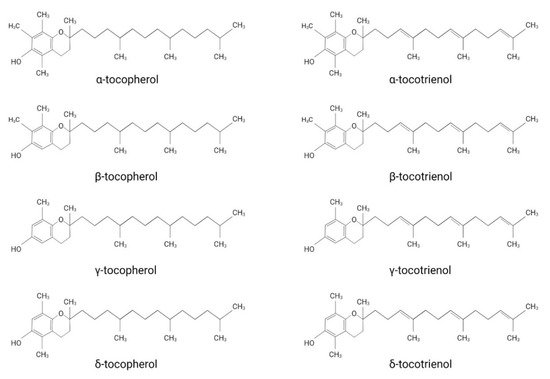
The definition of vitamin E and which derivates should be associated with this term are still under discussion. While vitamin E is extensively used as an umbrella term for different TP- and TT-forms, some authors use it as a synonym for α-TP. Azzi et al. justify the use of vitamin E as a synonym for α-TP by considering the classification of vitamins, which describes them as a group of substances essential for normal metabolism with deficiencies leading to disorders treatable by supplementation [4]. Accordingly, only α-TP should be named vitamin E, since it is the only form that has been shown to prevent the rare, inherited neurodegenerative disorder ataxia with vitamin E deficiency (AVED), which is caused by mutations in the gene encoding for α-tocopherol transfer protein ( TTPA ) [4][5]. However, since TPs and TTs are generally both implied when using the term vitamin E in the scientific literature, we will also use vitamin E as an umbrella term in this review.
Considering the consequences of vitamin E deficiency—ataxia, dysarthria and neuromuscular disorders [6]—it is clear that this substance plays an important role in the central and peripheral nervous systems. Of interest, abetalipoproteinemia, a disorder characterized by an inability to absorb fat and thereby profound the deficiency of chylomicrons, low-density-lipoprotein (LDL), and very-low-density lipoprotein (VLDL), all of which, being necessary for vitamin E absorption, result in ataxic neuropathy, retinal pigmentation, areflexia, and loss of proprioception [7]. Furthermore, equine neuroaxonal dystrophy/equine degenerative myeloencephalopathy, a neurodegenerative disorder affecting foals that resembles AVED, is associated with vitamin E deficiency, but is not associated with mutations in TTPA, such as is the case for AVED [8]. Interestingly, Kono et al. describe a case of juvenile spinocerebellar ataxia resulting from mutations in the phospholipid transfer protein ( PLTP ) gene, as well as TTPA [9].
Particularly in neurodegenerative disorders, vitamin E demonstrates notable benefits due to its antioxidant, anti-inflammatory and cholesterol-lowering properties [10]. Vitamin E supplementation as a therapy, particularly for neurodegenerative disorders, appears feasible and has been widely investigated, both in vitro and in vivo [11][12]. However, it has so far not been established in the prevention or treatment of these disorders, given the incoherent and sometimes contradictive results of interventional studies, and the apparent adverse effects of vitamin E supplementation [13]. Following recent insights into genetic polymorphisms, which play a crucial role in the metabolism of vitamin E, a new approach applying personalized medicine has emerged [14]. In this review, we will discuss the advantageous and disadvantageous effects of vitamin E in the context of neurodegenerative disease, as well as the factors that should be taken into consideration when tailoring personalized vitamin E supplementation strategies.
2. Genetics in the Metabolism of Vitamin E
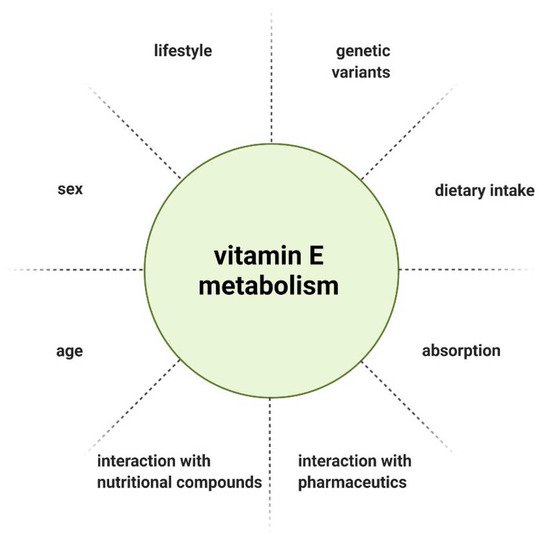
The metabolism and therefore bioavailability of vitamin E can be influenced by various factors, such as interaction with other nutritional compounds or pharmaceutics [15], gender [16], age [17], and lifestyle [18]. Due to the inhomogeneous outcome of interventional studies, individual response to vitamin E supplementation was considered as a potential explanation. Genetic heterogeneity arising from single nucleotide polymorphisms (SNPs) as a determinant of vitamin E homeostasis emerged as a hypothesis. SNPs are variations of a single nucleotide in a genome that can influence the biological properties of the encoded protein when occurring in coding regions [19][20]. Döring et al. described SNPs associated with genes that have a role in vitamin E metabolism, with the following genes considered as possible targets of SNPs, based on their function: lipoproteinlipase ( LPL ), tocopherol (α) transfer protein ( TTPA ), tocopherol-associated protein ( TAP ), multidrug resistance protein 2 ( MRP2 ), pregnane X receptor ( PXR ), and the genes encoding cytochrome P450 enzymes ( CYP3A5, CYP3A4, and CYP4F2 ). In the exons of TTPA, TAP and CYP3A5, only a few coding SNPs (cSNP) were found. The cSNP frequency calculated was 503–837 bp per cSNP and is therefore not highly polymorphic. There is also a common SNP in TAP, which leads to an exchange of amino acids in the N-terminal functional domain of the protein. In LPL, MRP2, PXR, CYP3A4, and CYP4F2, cSNPs were reported with a range of 100 bp per cSNP, constituting a high number of polymorphisms [21].
Several genome-wide and candidate gene association studies have since uncovered further SNPs in proteins involved in vitamin E absorption efficiency or catabolism. This includes SNPs in CYP4F2, the gene encoding for cytochrome P4504F2, which catabolizes vitamin E, as well as scavenger receptor class B member 1 ( SCARB1 ), which encodes scavenger receptor class B member 1, a plasma membrane receptor for high-density-lipoprotein (HDL), and the apolipoprotein A1/C3/A4/A5 -gene cluster, which encodes for apolipoproteins that are associated with α-TP status [22][23][24]. Borel et al. showed that the postprandial chylomicronic α-TP response to TP-rich meals was highly variable among subjects. The interindividual variability in TP bioavailability was estimated at 81%, to which 28 SNPs and 11 genes were identified to be potentially contributing. Since most vitamin E is transported from the intestine to the liver and other organs by chylomicrons, 7 genes were involved in the postprandial chylomicronic triacylglycerol response. The other 4 genes were associated with the chylomicronic-TP response. The authors further observed that the plasma TP-concentration was positively correlated with the chylomicronic α-TP response to TP-rich meals, highlighting the importance of interindividual ability to respond to dietary tocopherol intake as an influential factor for α-TP serum concentration [25].
α-TTP plays an important role in vitamin E homeostasis [26]. Resultant from two SNPs in the TTPA gene are two variants, E141K and R59W, which are associated with ataxia due to vitamin E deficiency. They cause the reduced binding of α-TP to α-TTP in vitro, as the variants are located near the ligand-binding domain of TTPA [27]. These findings are in line with Wright et al., who showed an approximate 3% lower baseline vitamin E plasma level associated with the TTPA (−980T > A) variant in the promotor region of TTPA [28]. Regarding vitamin E catabolism, cytochrome P450 plays a substantial role, as it catalyzes the initial step in the vitamin E-ω-hydroxylase pathway, and notably contributes to vitamin E levels [29]. Bardowell et al. discovered two SNPs differently influencing enzyme activity. While the CYP4F2 W12G variant leads to increased activity of the enzyme towards TPs and TTs, the CYP4F2 V433M variant reduces enzyme activity for TPs, but not for TTs [30]. In a clinical trial comparing these genetic variants, the V433M genotype was associated with significantly higher plasma α-TP levels after 48 weeks of vitamin E supplementation (pioglitazone versus vitamin E versus placebo for the treatment of non-diabetic patients with nonalcoholic steatohepatitis (PIVENS) study: r = 20.35, p = 0.004 and treatment of nonalcoholic fatty liver disease in children (TONIC) study: r = 20.34, p = 0.026) [31].
3. Vitamin E Can Fuel the Pathogenesis of Neurodegenerative Diseases
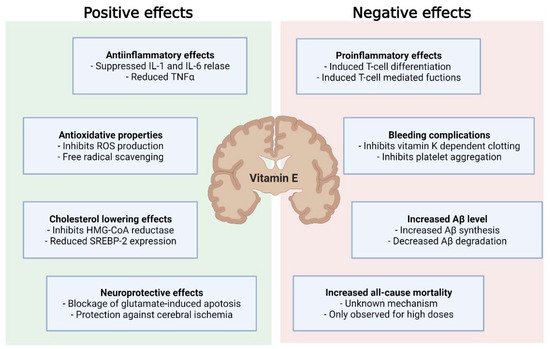
Although vitamin E displays beneficial effects, not only in neurodegenerative disease, but also cancer, cardiovascular disease and infections, current evidence does not support vitamin E supplementation in the treatment or prevention of these diseases. This is due to inconsistent study results and the evidence that high-dose vitamin E supplementation (>400 U/d) might increase all-cause mortality [13]. Since these studies often involve malnourished populations or combined vitamin E with other substances, it remains unclear whether or not this effect would be seen in a population that is not nutritionally deficient.
Mechanisms explaining increased all-cause mortality are still debated. However, a number of adverse effects have been observed for vitamin E. Very high dosages of vitamin E (44 mg/kg body weight) increased blood pressure in spontaneously hypertensive stroke prone (SHRSP) rats [32]. There was also a rise in: phosphorylated neurofilament H protein, a prognostic marker of neurological disorders [33] and acute ischemic stroke [34]; glial fibrillary acidic protein associated with AD [35]; and cathepsin D in the CNS [32]. A study investigating the neuroinflammatory response following ischemic stroke in α-TTP-deficient mice treated with an α-TP diet (1680 IU/d) observed an exacerbation of ischemic stroke injury, due to supraphysiological brain injury accompanied by an increase in markers of oxidative injury and neurodegeneration [36].
In contrast, an in vivo study using mice showed no effect of vitamin E on the expression of β-site of APP cleaving enzyme ( BACE-1 ) or a disintegrin and metalloproteinase domain-containing protein 10 ( ADAM-10 ) [37]. In another study, the same group was able to validate the findings of increased Aβ synthesis through vitamin E compound supplementation [38]. Here, α-TP and α-TT amplified Aβ synthesis after incubation on SH-SH5Y cells. These effects were mild. However, given the long preclinical phase of AD, even a small change in pathogenesis could result in earlier manifestation of clinical symptoms. To test the influence of α-TT on a non-AD in vitro model, Grimm et al. used SH-SY5Y WT cells. Surprisingly, there was an even more pronounced effect on Aβ synthesis [38]. A possible explanation could be the increased substrate presence in SH-SY5Y APP cells, resulting in enhanced Aβ degradation. Given that a high Aβ level sustained over a long period of time may increase Aβ catabolism, unaltered Aβ synthesis as observed by Arrozi et al. could be explained by the treatment period of over six months [39]. A study investigating TP levels in the human cortex observed an association between higher α- and γ-TP levels and lower total and activated microglia density in cortical regions, suggesting a microglia-mediated beneficial effect on the slowly accumulating AD neuropathology [40]. In later stages of AD, continued microglial activation can exacerbate tau pathology and negatively affect neurons and synapses [41][42]. During these stages, ameliorated microglial activation could be favorable. However, microglia activation may be protective in the early stages of AD, as microglia clear soluble Aβ, build protective barriers around Aβ plaques and remove debris [43][44]. Furthermore, a specific microglia cell type, disease-associated microglia (DAM), which has the potential to limit neurodegeneration, has been described [45]. Consequently, TPs being associated with reduced microglial activation highlights the possibility of further, perhaps adverse, effects of vitamin E supplementation on AD, as the transition into DAM might be restricted. On the contrary, in vitamin E-deficient mice, RNA-sequencing of the spinal cord demonstrated the upregulation of genes associated with the innate immune response, indicating that microglial activation may result from tocopherol deficiency [46].
Overall, this underpins the need for the proper timing of vitamin E supplementation in the course of neurodegenerative disease to improve the outcomes of treatment and mitigate adverse effects ( Figure 2 ).
4. Is Vitamin E Supplementation Suitable for Everyone?
Data from randomized controlled trials suggests that there are beneficial properties of vitamin E in neurodegenerative disease, as it is associated with a reduced risk of disease development and may slow disease progression [10]. However, due to inconsistent interventional trials that failed to validate the proposed favorable findings, vitamin E is not yet a part of the treatment or prevention of these disorders [47]. In addition, the evidence suggesting high-dose vitamin E may increase all-cause mortality warrants further caution [13][48]. Clearly, vitamin E is not suitable as a therapy that is to be used indiscriminately. However, in the current scientific landscape, the beneficial effects of vitamin E supplementation seem to outweigh the possible adverse effects, the latter to be considered for individual cases. As already discussed for AD, vitamin E has been shown to increase Aβ-synthesis in an in vitro AD model [38].
Due to its anti-inflammatory, antioxidative and cholesterol-lowering properties, vitamin E is considered a suitable therapeutic or preventive strategy. In line with the framework of personalized medicine, it could be argued that patients with a high inflammatory response, cholesterol levels or the occurrence of oxidative stress may benefit from vitamin E supplementation. In contrast, patients with low cholesterol, minimal inflammatory burden and little oxidative stress may be adversely affected by supplementation. Regarding the effects of α- and γ-TP on activated microglial cell density [40], supplementation can lead to either desirable or detrimental effects, dependent on disease progression and method of administration.
When analyzing a patient’s suitability for supplementation, not only metabolic status and disease progression need to be taken into consideration, but also comorbidities and comedication. Vitamin E inhibits vitamin K-dependent coagulation factors (II, VII, IX and X) in the presence of vitamin K deficiency, and thereby induces coagulopathy [49]. This indicates an increased bleeding risk in patients taking vitamin K-dependent anticoagulation. Considering that falls are common in the elderly [50] and patients with neurodegenerative diseases [51], hemorrhagic complications are of high importance.
Lastly, when evaluating vitamin E supplementation, the administered dose needs to be defined. There is no consistent recommendation for the daily intake of vitamin E, as the recommended dietary allowance (RDA) is defined by different methods across countries. Furthermore, various individual factors influence the metabolism and thereby bioavailability of vitamin E ( Figure 3 ) [15]. Considering the genetic polymorphisms in genes involved in the metabolism of vitamin E, the recommended intake should be tailored to the patient’s genetic profile.
References
- Evans, H.M.; Bishop, K.S. On the existence of a hitherto unrecognized dietary factor essential for reproduction. Science 1922, 56, 650–651.
- Torquato, P.; Marinelli, R.; Bartolini, D.; Giusepponi, D.; Cruciani, G.; Siragusa, L.; Galarini, R.; Sebastiani, B.; Gioiello, A.; Galli, F. Chapter 24—Vitamin E: Metabolism and molecular aspects. In Molecular Nutrition; Patel, V.B., Ed.; Academic Press: Cambridge, MA, USA, 2020; pp. 487–518. ISBN 978-0-12-811907-5.
- Chiroma, A.A.; Khaza’ai, H.; Abd Hamid, R.; Chang, S.K.; Zakaria, Z.A.; Zainal, Z. Analysis of Expression of Vitamin E-Binding Proteins in H2O2 Induced SK-N-SH Neuronal Cells Supplemented with α-Tocopherol and Tocotrienol-Rich Fraction. PLoS ONE 2020, 15, e0241112.
- Azzi, A. Many Tocopherols, One Vitamin E. Mol. Aspects Med. 2018, 61, 92–103.
- Azzi, A. Tocopherols, Tocotrienols and Tocomonoenols: Many Similar Molecules but Only One Vitamin E. Redox Biol. 2019, 26, 101259.
- Kemnic, T.R.; Coleman, M. Vitamin E Deficiency. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021.
- Muller, D.P.R. Vitamin E and Neurological Function. Mol. Nutr. Food Res. 2010, 54, 710–718.
- Finno, C.J.; Famula, T.; Aleman, M.; Higgins, R.J.; Madigan, J.E.; Bannasch, D.L. Pedigree Analysis and Exclusion of Alpha-Tocopherol Transfer Protein (TTPA) as a Candidate Gene for Neuroaxonal Dystrophy in the American Quarter Horse. J. Vet. Intern. Med. 2013, 27, 177–185.
- Kono, S.; Otsuji, A.; Hattori, H.; Shirakawa, K.; Suzuki, H.; Miyajima, H. Ataxia with Vitamin E Deficiency with a Mutation in a Phospholipid Transfer Protein Gene. J. Neurol. 2009, 256, 1180–1181.
- Bianchi, V.E.; Herrera, P.F.; Laura, R. Effect of Nutrition on Neurodegenerative Diseases. A Systematic Review. Nutr. Neurosci. 2019, 1–25.
- Schirinzi, T.; Martella, G.; Imbriani, P.; Di Lazzaro, G.; Franco, D.; Colona, V.L.; Alwardat, M.; Sinibaldi Salimei, P.; Mercuri, N.B.; Pierantozzi, M.; et al. Dietary Vitamin E as a Protective Factor for Parkinson’s Disease: Clinical and Experimental Evidence. Front. Neurol. 2019, 10, 148.
- Farina, N.; Llewellyn, D.; Isaac, M.G.E.K.N.; Tabet, N. Vitamin E for Alzheimer’s Dementia and Mild Cognitive Impairment. Cochrane Database Syst. Rev. 2017, 4, CD002854.
- Miller, E.R.; Pastor-Barriuso, R.; Dalal, D.; Riemersma, R.A.; Appel, L.J.; Guallar, E. Meta-Analysis: High-Dosage Vitamin E Supplementation May Increase All-Cause Mortality. Ann. Intern. Med. 2005, 142, 37–46.
- Galmés, S.; Serra, F.; Palou, A. Vitamin E Metabolic Effects and Genetic Variants: A Challenge for Precision Nutrition in Obesity and Associated Disturbances. Nutrients 2018, 10, 1919.
- Schmölz, L.; Birringer, M.; Lorkowski, S.; Wallert, M. Complexity of Vitamin E Metabolism. World J. Biol. Chem. 2016, 7, 14–43.
- Safari, T.; Miri, S.; Ghofran, O.; Fereidooni, F.; Niazi, A.A.; Bagheri, H.; Nematbakhsh, M. Gender Differences in Response to Vitamin E and C in Gentamicin Induced Nephrotoxicity in Wistar Rats. J. Nephropathol. 2017, 6, 338–345.
- Malik, A.; Eggersdorfer, M.; Trilok-Kumar, G. Vitamin E Status in Healthy Population in Asia: A Review of Current Literature. Int. J. Vitam. Nutr. Res. 2021, 91, 356–369.
- Alghadir, A.H.; Gabr, S.A.; Iqbal, Z.A.; Al-Eisa, E. Association of Physical Activity, Vitamin E Levels, and Total Antioxidant Capacity with Academic Performance and Executive Functions of Adolescents. BMC Pediatr. 2019, 19, 156.
- Wang, X.; Tomso, D.J.; Liu, X.; Bell, D.A. Single Nucleotide Polymorphism in Transcriptional Regulatory Regions and Expression of Environmentally Responsive Genes. Toxicol. Appl. Pharmacol. 2005, 207, 84–90.
- Zingg, J.-M.; Azzi, A.; Meydani, M. Genetic Polymorphisms as Determinants for Disease-Preventive Effects of Vitamin E. Nutr. Rev. 2008, 66, 406–414.
- Döring, F.; Rimbach, G.; Lodge, J.K. In Silico Search for Single Nucleotide Polymorphisms in Genes Important in Vitamin E Homeostasis. IUBMB Life 2004, 56, 615–620.
- Major, J.M.; Yu, K.; Chung, C.C.; Weinstein, S.J.; Yeager, M.; Wheeler, W.; Snyder, K.; Wright, M.E.; Virtamo, J.; Chanock, S.; et al. Genome-Wide Association Study Identifies Three Common Variants Associated with Serologic Response to Vitamin E Supplementation in Men. J. Nutr. 2012, 142, 866–871.
- Major, J.M.; Yu, K.; Wheeler, W.; Zhang, H.; Cornelis, M.C.; Wright, M.E.; Yeager, M.; Snyder, K.; Weinstein, S.J.; Mondul, A.; et al. Genome-Wide Association Study Identifies Common Variants Associated with Circulating Vitamin E Levels. Hum. Mol. Genet. 2011, 20, 3876–3883.
- Ferrucci, L.; Perry, J.R.B.; Matteini, A.; Perola, M.; Tanaka, T.; Silander, K.; Rice, N.; Melzer, D.; Murray, A.; Cluett, C.; et al. Common Variation in the Beta-Carotene 15,15′-Monooxygenase 1 Gene Affects Circulating Levels of Carotenoids: A Genome-Wide Association Study. Am. J. Hum. Genet. 2009, 84, 123–133.
- Borel, P.; Desmarchelier, C.; Nowicki, M.; Bott, R.; Tourniaire, F. Can Genetic Variability in α-Tocopherol Bioavailability Explain the Heterogeneous Response to α-Tocopherol Supplements? Antioxid. Redox Signal. 2015, 22, 669–678.
- Peltzer, R.M.; Kolli, H.B.; Stocker, A.; Cascella, M. Self-Assembly of α-Tocopherol Transfer Protein Nanoparticles: A Patchy Protein Model. J. Phys. Chem. B 2018, 122, 7066–7072.
- Bromley, D.; Anderson, P.C.; Daggett, V. Structural Consequences of Mutations to the α-Tocopherol Transfer Protein Associated with the Neurodegenerative Disease Ataxia with Vitamin E Deficiency. Biochemistry 2013, 52, 4264–4273.
- Wright, M.E.; Peters, U.; Gunter, M.J.; Moore, S.C.; Lawson, K.A.; Yeager, M.; Weinstein, S.J.; Snyder, K.; Virtamo, J.; Albanes, D. Association of Variants in Two Vitamin e Transport Genes with Circulating Vitamin e Concentrations and Prostate Cancer Risk. Cancer Res. 2009, 69, 1429–1438.
- Parker, R.S.; Sontag, T.J.; Swanson, J.E.; McCormick, C.C. Discovery, Characterization, and Significance of the Cytochrome P450 Omega-Hydroxylase Pathway of Vitamin E Catabolism. Ann. N. Y. Acad. Sci. 2004, 1031, 13–21.
- Bardowell, S.A.; Stec, D.E.; Parker, R.S. Common Variants of Cytochrome P450 4F2 Exhibit Altered Vitamin E--Hydroxylase Specific Activity. J. Nutr. 2010, 140, 1901–1906.
- Athinarayanan, S.; Wei, R.; Zhang, M.; Bai, S.; Traber, M.G.; Yates, K.; Cummings, O.W.; Molleston, J.; Liu, W.; Chalasani, N. Genetic Polymorphism of Cytochrome P450 4F2, Vitamin E Level and Histological Response in Adults and Children with Nonalcoholic Fatty Liver Disease Who Participated in PIVENS and TONIC Clinical Trials. PLoS ONE 2014, 9, e95366.
- Miyamoto, K.; Shiozaki, M.; Shibata, M.; Koike, M.; Uchiyama, Y.; Gotow, T. Very-High-Dose α-Tocopherol Supplementation Increases Blood Pressure and Causes Possible Adverse Central Nervous System Effects in Stroke-Prone Spontaneously Hypertensive Rats. J. Neurosci. Res. 2009, 87, 556–566.
- Lee, Y.; Lee, B.H.; Yip, W.; Chou, P.; Yip, B.-S. Neurofilament Proteins as Prognostic Biomarkers in Neurological Disorders. Curr. Pharm. Des. 2019, 25, 4560–4569.
- Hutanu, A.; Maier, S.; Bălaşa, R.; Oprea, O.; Barbu, S.; Septimiu, V.; Dobreanu, M. Plasma Phosphorylated Neurofilament Heavy Chains as a Potential Marker for Ischemic Stroke Patients. Rev. Romana Med. Lab. 2018, 26, 59–64.
- Chatterjee, P.; Pedrini, S.; Stoops, E.; Goozee, K.; Villemagne, V.L.; Asih, P.R.; Verberk, I.M.W.; Dave, P.; Taddei, K.; Sohrabi, H.R.; et al. Plasma Glial Fibrillary Acidic Protein Is Elevated in Cognitively Normal Older Adults at Risk of Alzheimer’s Disease. Transl. Psychiatry 2021, 11, 1–10.
- Khanna, S.; Heigel, M.; Weist, J.; Gnyawali, S.; Teplitsky, S.; Roy, S.; Sen, C.K.; Rink, C. Excessive α-Tocopherol Exacerbates Microglial Activation and Brain Injury Caused by Acute Ischemic Stroke. FASEB J. 2015, 29, 828–836.
- Gaedicke, S.; Zhang, X.; Huebbe, P.; Boesch-Saadatmandi, C.; Lou, Y.; Wiswedel, I.; Gardemann, A.; Frank, J.; Rimbach, G. Dietary Vitamin E, Brain Redox Status and Expression of Alzheimer’s Disease-Relevant Genes in Rats. Br. J. Nutr. 2009, 102, 398–406.
- Grimm, M.O.W.; Regner, L.; Mett, J.; Stahlmann, C.P.; Schorr, P.; Nelke, C.; Streidenberger, O.; Stoetzel, H.; Winkler, J.; Zaidan, S.R.; et al. Tocotrienol Affects Oxidative Stress, Cholesterol Homeostasis and the Amyloidogenic Pathway in Neuroblastoma Cells: Consequences for Alzheimer’s Disease. Int. J. Mol. Sci. 2016, 17, 1809.
- Pahrudin Arrozi, A.; Shukri, S.N.S.; Wan Ngah, W.Z.; Mohd Yusof, Y.A.; Ahmad Damanhuri, M.H.; Jaafar, F.; Makpol, S. Comparative Effects of Alpha- and Gamma-Tocopherol on Mitochondrial Functions in Alzheimer’s Disease In Vitro Model. Sci. Rep. 2020, 10, 8962.
- de Leeuw, F.A.; Schneider, J.A.; Agrawal, S.; Leurgans, S.E.; Morris, M.C. Brain Tocopherol Levels Are Associated with Lower Activated Microglia Density in Elderly Human Cortex. Alzheimer Dement. 2020, 6, e12021.
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.-M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M.-Y. Synapse Loss and Microglial Activation Precede Tangles in a P301S Tauopathy Mouse Model. Neuron 2007, 53, 337–351.
- Salter, M.W.; Stevens, B. Microglia Emerge as Central Players in Brain Disease. Nat. Med. 2017, 23, 1018–1027.
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s Disease. J. Cell Biol. 2018, 217, 459–472.
- Kaur, D.; Sharma, V.; Deshmukh, R. Activation of Microglia and Astrocytes: A Roadway to Neuroinflammation and Alzheimer’s Disease. Inflammopharmacology 2019, 27, 663–677.
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17.
- Finno, C.J.; Bordbari, M.H.; Gianino, G.; Ming-Whitfield, B.; Burns, E.; Merkel, J.; Britton, M.; Durbin-Johnson, B.; Sloma, E.A.; McMackin, M.; et al. An Innate Immune Response and Altered Nuclear Receptor Activation Defines the Spinal Cord Transcriptome during Alpha-Tocopherol Deficiency in Ttpa-Null Mice. Free Radic. Biol. Med. 2018, 120, 289–302.
- Lloret, A.; Esteve, D.; Monllor, P.; Cervera-Ferri, A.; Lloret, A. The Effectiveness of Vitamin E Treatment in Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 879.
- Chai, W.; Maskarinec, G.; Franke, A.A.; Monroe, K.R.; Park, S.-Y.; Kolonel, L.N.; Wilkens, L.R.; Le Marchand, L.; Cooney, R.V. Association of Serum γ-Tocopherol Levels with Mortality: The Multiethnic Cohort Study. Eur. J. Clin. Nutr. 2020, 74, 87–96.
- Corrigan, J.J. The Effect of Vitamin E on Warfarin-Induced Vitamin K Deficiency. Ann. N. Y. Acad. Sci. 1982, 393, 361–368.
- Haagsma, J.A.; Olij, B.F.; Majdan, M.; van Beeck, E.F.; Vos, T.; Castle, C.D.; Dingels, Z.V.; Fox, J.T.; Hamilton, E.B.; Liu, Z.; et al. Falls in Older Aged Adults in 22 European Countries: Incidence, Mortality and Burden of Disease from 1990 to 2017. Inj. Prev. 2020, 26, i67–i74.
- Keleman, A.; Wisch, J.K.; Bollinger, R.M.; Grant, E.A.; Benzinger, T.L.; Morris, J.C.; Ances, B.M.; Stark, S.L. Falls Associate with Neurodegenerative Changes in ATN Framework of Alzheimer’s Disease. J. Alzheimer Dis. 2020, 77, 745–752.