Monoamine oxidase (E.C. 1.4.3.4) enzymes MAO A and MAO B are FAD-containing proteins located on the outer face of the mitochondrial inner membrane, retained there by hydrophobic interactions and a transmembrane helix. The redox co-factor (FAD) is covalently attached to a cysteine and buried deep inside the protein behind an aromatic cage that aligns substrates towards the flavin. MAO metabolizes neurotransmitters such as dopamine and serotonin in the nervous system so is a target for drugs to modify amine levels. MAO also metabolizes a wide range of biogenic amines in all tissues. Current accumulated evidence, particularly from theoretical modelling, supports hydride transfer as the chemical mechanism. The long active site cavity accommodates a wide chemical variety of small molecules designed as inhibitors, including coumarins, chromones, triazoles, and more. Inactivators that bind covalently to MAO include hydrazines, cyclopropylamines and propargylamines. This entry is an extract adapted from a review outlining the remaining uncertainties in the understanding of this key drug target.
- chemical mechanism
- kinetic mechanism
- oxidation
- protein flexibility
1. Introduction
This brief overview of the chemical enzymology of monoamine oxidase (MAO, E.C. 1.4.3.4) is an extract adapted from a recent review outlining the remaining uncertainties in the understanding of this key drug target [1]. The monoamine oxidase enzymes MAO A and MAO B are FAD-containing proteins located on the outer face of the mitochondrial inner membrane, retained there by hydrophobic interactions and a transmembrane helix [2][3][4][5]. The redox co-factor (FAD) is covalently attached to a cysteine and buried deep inside the protein [6]. Two human genes code for MAO A and MAO B that have about 70% sequence identity and are regulated separately resulting in varied proportions in different cell types [7][8]. MAO metabolizes neurotransmitters such as dopamine and serotonin in the nervous system and a wide range of biogenic amines in all tissues [9]. Inhibition of MAO alters the amine balance in the brain so many classes of molecules have been explored in the search for antidepressants and drugs to combat neurodegeneration. Examples of substrates and inhibitors and their kinetic parameters are given in Table 1.
Table 1. Some substrates, inhibitors and inactivators of MAO with their kinetic parameters.
|
MAO A |
Compound |
MAO B |
|||
|
KM |
Vmax |
Substrates A |
|
KM |
Vmax |
|
137 |
228 |
Serotonin |
1093 |
6.6 |
|
|
212 |
680 |
Dopamine |
229 |
702 |
|
|
140 |
20 |
2-Phenylethylamine |
4 |
309 |
|
|
127 |
182 |
Tyramine |
107 |
343 |
|
|
Ki |
|
Reversible |
|
Ki |
|
|
18 |
|
D-Amphetamine |
250 |
|
|
|
3 |
|
MPP+ c |
230 |
|
|
|
365 |
|
Safinamide |
0.45 |
|
|
|
11.5 |
|
Moclobemide |
>100 |
|
|
|
0.005 |
|
Harmine |
121 |
|
|
|
0.028 |
|
Methylene Blue |
5.5 |
|
|
|
KI |
kinact |
Irreversible |
|
KI |
kinact |
|
3.1 |
0.12 |
Phenelzine |
50 a |
0.9 a |
|
|
7.7 |
0.78 |
Tranylcypromine |
0.7 |
0.16 |
|
|
193 |
0.25 |
Selegiline d |
0.1 |
0.53 |
|
|
9.7 |
0.007 |
Rasagiline b |
0.7 |
0.05 |
|
|
0.03 |
1.8 |
Clorgyline |
1.8 |
0.02 |
|
|
3.0 |
0.39 |
Contilisant e |
4.6 |
0.19 |
|
Data: A from [10]; B summarized in [11] from references therein; C from [12] except a [13] and b [14]. c MPP+ is 1-methyl-4-phenyl-pyridinium; d Selegiline is also known as L-deprenyl; e Contilisant is a multi-target compound which also inhibits cholinesterase and binds to the histamine H3 receptor [15].
MAO became an important drug target when a tuberculosis drug was found to improve the mood of patients. The large antidepressant market fueled the discovery of other irreversible inhibitors of MAO [16][17][18] that act by covalent attachment [19][20][21]. The common drugs such as selegiline (Table 1, used in the treatment of Parkinson’s disease) modify the flavin, but binding to an amino acid residue was also reported for some compounds [22][23]. The major side-effect of treatment with the irreversible inhibitors is the tyramine-induced pressor “cheese effect” due to the inhibition of intestinal MAO A that normally metabolizes dietary tyramine found at high levels in aged cheeses [24]. Drug discovery for MAO inhibition then switched to reversible inhibitors, although the selective irreversible inactivators of MAO still remain important in the treatment of neurodegenerative diseases. MAO inhibitors (MAOI) can conserve the decreased levels of monoamine neurotransmitters in a deteriorating brain. For the complex pathology of neurodegeneration, the concept of multi-target drugs has driven design of a variety of compounds incorporating MAO inhibition, for example rasagiline and contilisant (Table 1) [14][15]. Both of these compounds incorporate the propargylamine fragment that gives mechanism-based inactivation of the enzyme [12].
Although it is almost 100 years after the identification of monoamine oxidase, 70 years after its adoption as a drug target and 20 years after its structure was solved, some interesting questions still remain to be explored (see [1]. This entry provides lead-in references for details of the research in key areas of structure, mechanism, kinetics and selectivity relevant for drug design.
2. Structure
High-resolution structures are key tools in modern enzymology and drug design. Docking and molecular dynamics inform improvement of drug candidate affinity and, more recently, have led to quantum mechanical models of the enzyme reaction. MAO was originally extracted from the mitochondrial inner membranes of liver (MAO B) or placenta (MAO A) using phospholipases to digest the surrounding lipids and high concentrations of Triton to solubilize the dimeric protein [25]. The breakthrough for structural work was the cloning and high-level expression of the human genes in Picchia pastoris [26][27]. Isolation of purified MAO requires specific detergents, care to avoid oxidation of the 8 free cysteine thiols, and addition of a ligand to stabilize the protein. Indeed, the crystal structures have ligand bound to the active site [4][5][21]. The structures best suited to docking are those with non-covalently bound inhibitors because the covalent inactivators such as selegiline (L-deprenyl) or clorgyline form the covalent bond with the reduced flavin [28]. Careful preparation of the structures, including re-insertion of the cysteine-flavin bond before use for docking, molecular dynamics or quantum mechanical modeling is essential [29][30][31][32][33][34][35].
The structures of the MAO enzymes are well-described in reviews [36][37][38]. The terminal 27 residues form an alpha helix embedded in the membrane but interactions with the membrane phospholipids are also important. The membrane association influences the catalytic properties, in particular the substrate KM [39][40], perhaps because the entrance to the substrate cavity is close to the membrane surface. In both MAO A and MAO B the hydrophobic substrate cavity leads to an aromatic cage of tyrosines that aligns the substrate towards the N5 of the FAD where catalysis takes place. Figure 1 shows a schematic view of the active site of MAO B. The differences in substrate and inhibitor specificity between MAO A and MAO B come from a few residues that differ, providing distinct interactions influenced by shape and hydrogen bonding (see below in section 4, Selectivity and drug design). In particular, human MAO B features a constriction in the active site whereas MAO A has a wider single cavity.
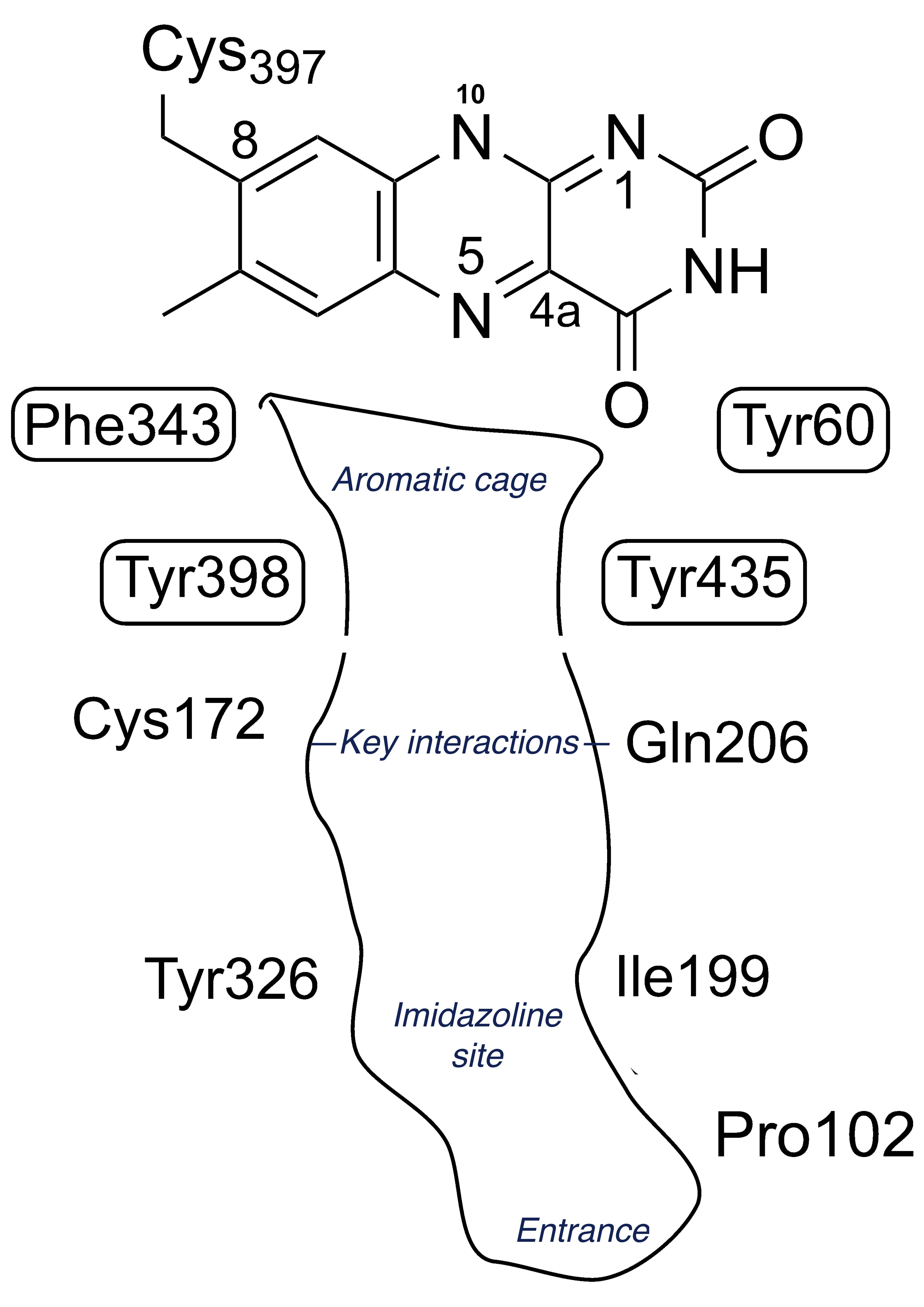
Figure 1. Schematic view of the cysteinyl-flavin and key amino acids in the active site of MAO B.
3. Chemical Mechanism
Despite the overall similarity in structure, MAO A and MAO B have different substrate and inhibitor selectivities (for example, [10][41][42][43] defined by the distinct hydrophobicity and binding site architectures but the key region close to the FAD is the same [43]. The chemical mechanism was substantially explored by traditional chemical approaches in the 1990s. Four mechanisms for oxidation of the amine by MAO have attracted investigation: single electron transfer (SET), the polar nucleophilic mechanism or (as found in some other FAD-dependent oxidases) hydride extraction, and a derivative thereof, two-step hydride transfer (see Figure 2). The strong chemical evidence for the single electron transfer mechanism came from in depth studies on the inactivation of MAO by cyclopropylamines and remains a probable mechanism for conversion of a cyclopropylamine into an activated product capable of forming a covalent bond either to the FAD or to a cysteine [22][44]. However, inability to detect a flavin semiquinone radical by electron paramagnetic resonance (EPR) during substrate turnover cast doubt on SET as the general catalytic pathway. Quantum chemical calculations for a SET mechanism for para-substituted benzylamines gave inverse correlation with experimental data, confirming that this is not the mechanism of substrate oxidation [45].
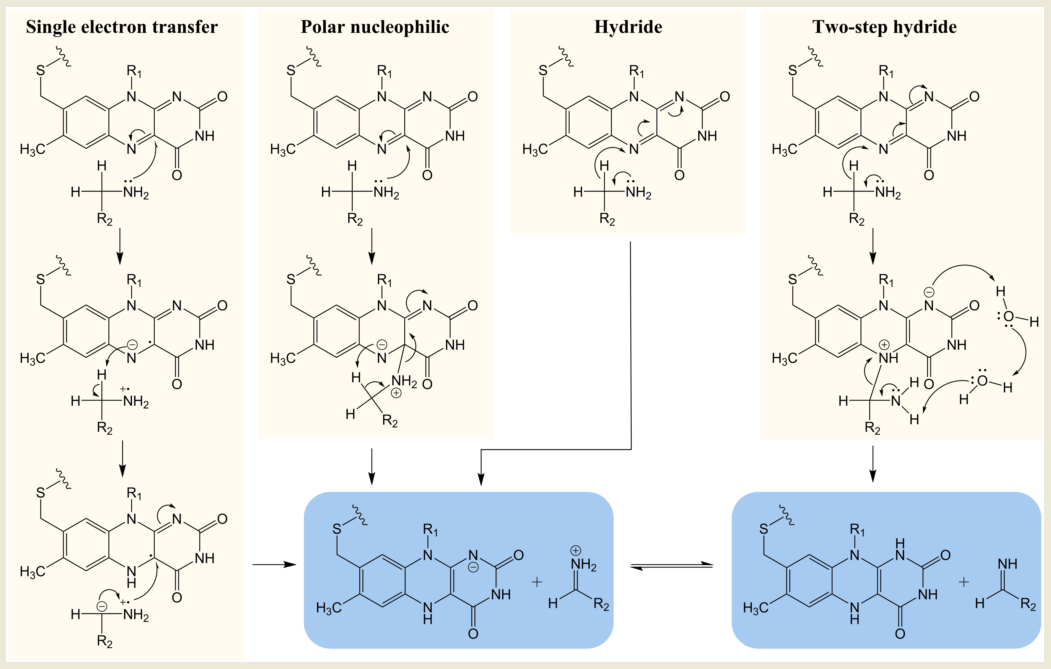
Figure 2. Chemical mechanisms proposed for MAO-catalyzed oxidation of amines.
In depth studies based on Hammett correlations were carried out in support of a polar nucleophilic mechanism for MAO A where a convincing positive correlation of oxidation rate with electronic effects of benzylamine substituents was obtained (ρ = 2)[46]. In this study with MAO A, the more electron withdrawing character of the substituent, the faster was the rate of oxidation, consistent with the polar nucleophilic mechanism whereby a transient C4a adduct was formed with the flavin. In contrast, for MAO B, the substituents that more efficiently stabilize the developing electron density in the transition state slowed the rate, prompting the suggestion that MAO A and MAO B followed different mechanisms [47]. The slower rate of MAO B with electron withdrawing substituents could be consistent with the hydride mechanism, but nitrogen kinetic isotope effects for the oxidation of benzylamine by MAO B showed that the rehybridization of the N(5) nitrogen atom required for reduction of the flavin and the cleavage of the CH bond for oxidation of the amine were not concerted [48]. A quantum mechanics/molecular mechanics (QM/MM) study of benzylamine oxidation by the MAO also suggested that the transfer of electrons and proton was asynchronous, supporting the polar nucleophilic mechanism [49].
Hydride transfer was established for D-amino acid oxidase after high-resolution structures with bound substrates were solved giving insight into the probable transition state [50]. However, X-ray structures of MAO are available only with inhibitors bound (see, for example, [36]), but since computational modeling based on these structures became practical, evidence for a hydride transfer reaction in both MAO A and MAO B has been mounting [34][51][52][53]. In a QM/MM simulation of the rate-limiting step for the serotonin oxidation via hydride transfer to the N5 of the FAD in MAO A, the calculated barrier was 14.8 ± 0.8 kcal mol−1, in good agreement with the experimental value of 16.0 kcal mol−1. The two-step hydride transfer mechanism shown for MAO B shown in Figure 2 proposes hydride abstraction from the substrate and relaxation of the resulting flavin-substrate adduct into a neutral amine and fully reduced flavin. The second part of the reaction is presumably facilitated by two water molecules present at the binding site [53]. The activation free energy for the hydride transfer in this computational study was shown to be roughly two-times smaller (24 kcal mol−1) than for any other oxidation mechanism by MAO B.
3. Kinetic mechanism and redox implications
Early work varying both benzylamine and oxygen indicated a ping-pong mechanism for rat MAO [54][55], but later kinetic studies demonstrated that some substrates followed a ternary complex mechanism (Figure 3) [56][57][58]. Spontaneous hydrolysis of the imine product to the corresponding aldehyde and ammonia, at least some of which might occur in the active site, complicates the picture [59]. However, hydrolysis of imine in the active site is probably not the prevailing mechanism, but rather, the imine is primarily hydrolyzed non-enzymatically outside the enzyme [60]. The number (and orientation) of water molecules in the active site depends on the particular isozyme (MAO A versus MAO B) and the nature of the substrate [61][62][63]. Many water molecules can be of structural type (e.g., involved in hydrogen bonding), making them very unlikely to take part in any type of reaction [64]. From results using a benzylamine analog oxidized to a spectrally distinct imine product, it has been argued that the reduced enzyme-imine complex is reoxidized before the bound imine signal is lost [59]. If either ammonia or imine remained bound, free reduced enzyme would be minimal, and no new ligand could bind. However, the kinetics demonstrating binding of inhibitor to reduced enzyme indicate that free reduced enzyme is present during turnover [56][57][65].
Ultimately, whether a pool of free reduced enzyme builds up during turnover will depend on the rate constants for the half-reactions, the concentrations of the substrates (amine and oxygen), and the affinities of the oxidized (Eo) and reduced (Er) forms of the enzyme for their ligands (see Figure 3). Although reduction of the FAD by the amine substrate is the slowest step in the reaction catalyzed by MAO, there are three possible pathways for the reoxidation of the reduced FAD co-factor. The rate of reoxidation of the reduced MAO B without ligand is slow (kox, 1.8 s−1 at 0.28 mM oxygen) and whether Ered-P is oxidized before dissociation of the imine product is questioned. When some substrates are preincubated with reduced MAO, the rate of oxidation increases 2–100 times [58]; so for those substrates, the pathway in the green box dominates. Based on data from [58], 2-phenylethylamine (PEA) reduces bovine MAO B at 572 s−1 (kred), rapidly generating Ered, but the steady-state turnover rate is 3.6 s−1. The imbalance in rates means that the MAO is mostly reduced (Ered) during steady-state oxidation of PEA. Based on steady-state and half-reaction studies, oxidation of PEA is mainly via the binary pathway. With benzylamine as substrate, the reduction and reoxidation rates are similar (10.9 and 7.6 s−1, respectively), and reoxidation is mainly via the ternary complex pathway indicated by the green rectangle. It should be noted that reoxidation rates are influenced by the concentration of oxygen ([O2]). Even with these similar rates of reduction and oxidation, some intermediate Ered is available for binding of inhibitor as has been demonstrated for both bovine MAO B [56] and human MAO B [65]. That the affinities of the oxidized (Eo) and reduced (Er) forms of the enzyme for inhibitors can differ was demonstrated for the tricyclic antidepressant amitriptyline which gives a Ki of 0.5 mM with Eo but 2.5 mM with Er [66]. The question is important for the drug design process because IC50 measurements used to screen new compounds will vary with the proportion of reduced enzyme present during turnover which depends upon the assay and substrate used [65][66].
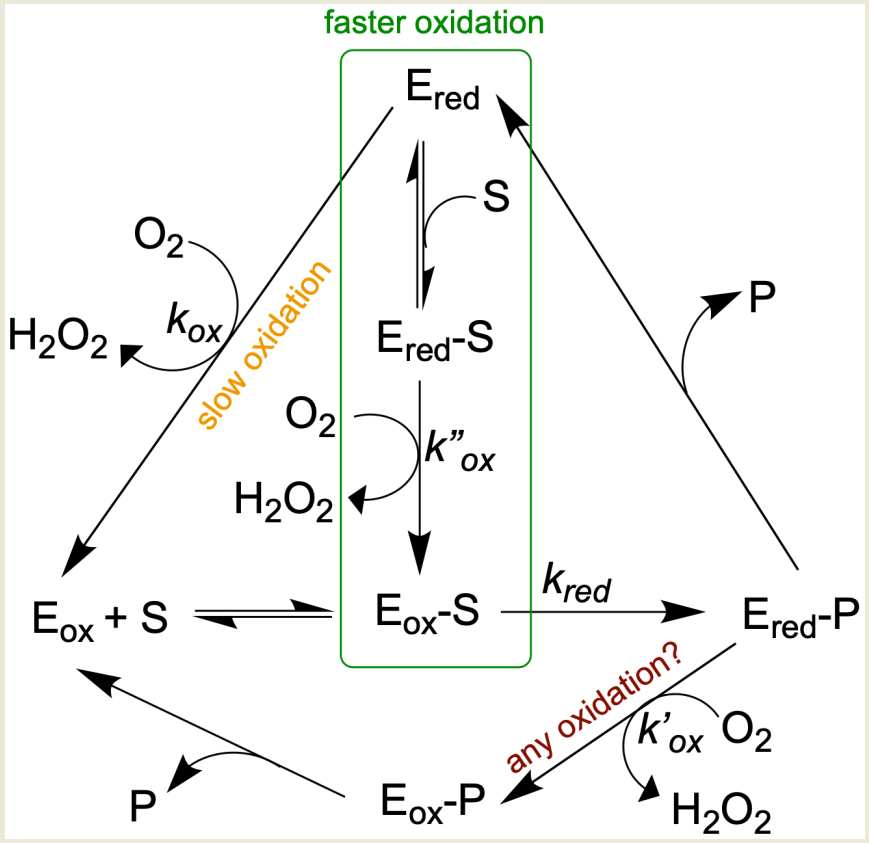
Figure 3. Alternative pathways for oxidation complicate MAO kinetic studies.
4. Selectivity and drug design
The rate of catalysis by MAO is slow (3–10 s−1 [58]) but specific (the pro-R hydrogen from the α carbon next to the amine is removed [67]). Compared with MAO A, the tighter substrate cavity of MAO B results in increased steric hindrance and in distinctive amino acid residue interactions with small molecules. This makes MAO B more sensitive to the absolute configuration at chiral centers of enantiomeric substrates and inhibitors [42]. Specificity is also governed by the hydrophobic and hydrophilic regions of the cavity. The environments of both substrate cavities are generally hydrophobic, but MAO B is slightly more so. Consequently, by transforming some regular MAO B inhibitors into quaternary ammonium salts, selective MAO A inhibitors were obtained [68]. Within each cavity, docking of variants within the many classes of inhibitors has demonstrated the importance of specific interactions such as hydrogen bonding. Specially, MAO A Phe208-Ile335 and MAO B Ile199-Tyr326 pairs were suggested to be the major determinants that govern the differential inhibitor specificities of the two isozymes [39]. Site-directed mutagenesis studies showed that Ile335 in MAO A and Tyr326 in MAO B are key amino acid residues determining substrate and inhibitor specificities in human and rat MAO A and B [3][69][70]. However, the wide range of effective inhibitors demonstrates that affinity for a ligand is determined by multiple interactions within the large cavity (for examples, see recent reviews focussed on inhibitors: [71][72][73]. Perhaps of interest for applications in biological tissues or in vivo is the observation that geometric isomers of cis- and trans-1-propargyl-4-styrylpiperidine distinguish between MAO A and MAO B. The cis isomers are potent human MAO A irreversible inhibitors whereas the trans analogues inactivate only MAO B both in vitro and in mouse brain [74].
Although MAO A is the target for antidepressants, MAO B is the key target in maintaining amine levels in deteriorating brain. Selegiline inactivates MAO B but can also inactivate MAO A at higher doses or with prolonged use, so transdermal administration was adopted to avoid inactivating MAO A in the gut where it is essential to protect from dietary biogenic amines. More recently, the reversible inhibitor safinamide has been found to be effective although selegiline remains most common in the treatment of Parkinson’s disease to delay or reduce the need for l-dopa [75]. However, a meta-analysis comparing MAO inhibitors, either alone or with l-dopa, showed that selegiline was the most effective drug and that both it and rasagiline were more effective than safinamide (selegiline > rasagiline > safinamide) [76].
The question of whether reversible or irreversible inhibition provides the better pharmacological intervention remains under debate. The effect of a successful drug is governed by binding affinity and by pharmacokinetics. For a high affinity reversible inhibitor, side effects could be low, and the inhibition is easily reversed. Indeed, it might be desirable that a surge in the amine substrate concentration would out-compete the inhibitor. For irreversible inhibitors, reaching a steady-state level of inhibition might take longer if the dose is low, but the effect would persist until the inactivated enzyme was replaced by a new protein molecule (a slow process for MAO) so that irreversible inhibitors could provide more stable levels of inhibition in patients. To achieve the desired clinical effect in depression or Parkinson’s disease, MAO inhibition must be greater than 75% [77]. For reversible inhibitors, maintaining high levels of inhibition depends on regular administration and requires high affinity, so perhaps the slow wash-out of the irreversible inhibitors is an advantage.
Overall, drug design remains the major opportunity for treatment of depression, neurodegeneration or cancer. Identification of key interactions at the catalytic site and appreciation of the chemical and kinetic complexities is vital for research and innovation towards new lines of MAO inhibiting drugs with enhanced selectivity and potency.
References
- Ramsay, R. R.; Albreht, A., Questions in the Chemical Enzymology of MAO. Chemistry 2021, 3, (3), 959-978.
- Binda, C.; Edmondson, D. E.; Mattevi, A., 1OJC: Human Monoamine Oxidase B In Complex With N-(2-Aminoethyl)-P-Chlorobenzamide. Worldwide Protein Data Bank 2003.
- Ma, J. C.; Yoshimura, M.; Yamashita, E.; Nakagawa, A.; Ito, A.; Tsukihara, T., Structure of rat monoamine oxidase A and its specific recognitions for substrates and inhibitors. Journal of Molecular Biology 2004, 338, (1), 103-114.
- De Colibus, L.; Li, M.; Binda, C.; Lustig, A.; Edmondson, D. E.; Mattevi, A., Three-dimensional structure of human monoamine oxidase A (MAO A): Relation to the structures of rat MAO A and human MAO B. Proceedings of the National Academy of Sciences of the United States of America 2005, 102, (36), 12684-12689.
- Son, S. Y.; Ma, A.; Kondou, Y.; Yoshimura, M.; Yamashita, E.; Tsukihara, T., Structure of human monoamine oxidase A at 2.2-angstrom resolution: The control of opening the entry for substrates/inhibitors. Proceedings of the National Academy of Sciences of the United States of America 2008, 105, (15), 5739-5744.
- Edmondson, D. E.; Binda, C.; Mattevi, A., The FAD binding sites of human monoamine oxidases A and B. NeuroToxicology 2004, 25, (1-2), 63-72.
- Shih, J. C.; Chen, K.; Ridd, M. J., Monoamine oxidase: From genes to behavior. Annual Review of Neuroscience 1999, 22, 197-217.
- Shih, J. C., Monoamine oxidase isoenzymes: genes, functions and targets for behavior and cancer therapy. Journal of Neural Transmission 2018, 125, (11), 1553-1566.
- Tipton, K. F., 90years of monoamine oxidase: some progress and some confusion. Journal of Neural Transmission 2018, 125, (11), 1519-1551.
- Youdim, M. B. H.; Edmondson, D.; Tipton, K. F., The therapeutic potential of monoamine oxidase inhibitors. Nature Reviews Neuroscience 2006, 7, (4), 295-309.
- Ramsay, R. R., Molecular aspects of the activity and inhibition of the FAD-containing monoamine oxidases. In Pharmaceutical Biocatalysis: Fundamentals, Enzyme Inhibitors, and Enzymes in Health and Diseases., Grunwald, P., Ed. Pan Stanford Publishing Pte. Ltd.,: Singapore, 2019; Vol. 4.
- Ramsay, R. R. B., L.; Maniquet, A.; Hagenow, S.; Pappalardo, M.; Saija, M.C.; Bryant, S.D.; Albreht, A.; Guccione, S. Molecules 2020, 25, 5908. , Parameters for irreversible inactivation of monoamine oxidase. In Molecules, MDPI: 2020; Vol. 25.
- Binda, C.; Wang, J.; Li, M.; Hubalek, F.; Mattevi, A.; Edmondson, D. E., Structural and mechanistic studies of arylalkylhydrazine inhibition of human monoamine oxidases A and B. Biochemistry 2008, 47, (20), 5616-5625.
- Hubalek, F.; Binda, C.; Li, M.; Herzig, Y.; Sterling, J.; Youdim, M. B. H.; Mattevi, A.; Edmondson, D. E., Inactivation of purified human recombinant monoamine oxidases A and B by rasagiline and its analogues. Journal of Medicinal Chemistry 2004, 47, (7), 1760-1766.
- Bautista-Aguilera, O. M.; Hagenow, S.; Palomino-Antolin, A.; Farre-Alins, V.; Ismaili, L.; Joffrin, P. L.; Jimeno, M. L.; Soukup, O.; Janockova, J.; Kalinowsky, L.; Proschak, E.; Iriepa, I.; Moraleda, I.; Schwed, J. S.; Martinez, A. R.; Lopez-Munoz, F.; Chioua, M.; Egea, J.; Ramsay, R. R.; Marco-Contelles, J.; Stark, H., Multitarget-Directed Ligands Combining Cholinesterase and Monoamine Oxidase Inhibition with Histamine H3R Antagonism for Neurodegenerative Diseases. Angewandte Chemie-International Edition 2017, 56, (41), 12765-12769.
- Kim, T.; Xu, C.; Amsterdam, J. D., Relative effectiveness of tricyclic antidepressant versus monoamine oxidase inhibitor monotherapy for treatment-resistant depression. Journal of Affective Disorders 2019, 250, 199-203.
- Mann, J. J.; Aarons, S. F.; Wilner, P. J.; Keilp, J. G.; Sweeney, J. A.; Pearlstein, T.; Frances, A. J.; Kocsis, J. H.; Brown, R. P., A controlled-study of the antidepressant efficacy and side-effects of (-)-deprenyl - a selective monoamine-oxidase inhibitor. Archives of General Psychiatry 1989, 46, (1), 45-50.
- Quitkin, F.; Rifkin, A.; Klein, D. F., Mono-amine oxidase-inhibitors - review of anti-depressant effectiveness. Archives of General Psychiatry 1979, 36, (7), 749-760.
- Maycock, A. L.; Abeles, R. H.; Salach, J. I.; Singer, T. P., Structure of covalent adduct formed by interaction of 3-dimethylamino-1-propyne and flavin of mitochondrial amine oxidase. Biochemistry 1976, 15, (1), 114-125.
- Gartner, B.; Hemmerich, P.; Zeller, E. A., Structure of flavin adducts with acetylenic substrates - Chemistry of monoamine-oxidase and lactate oxidase Inhibition. European Journal of Biochemistry 1976, 63, (1), 211-221.
- Binda, C.; Newton-Vinson, P.; Hubalek, F.; Edmondson, D. E.; Mattevi, A., Structure of human monoamine oxidase B, a drug target for the treatment of neurological disorders. Nature Structural Biology 2002, 9, (1), 22-26.
- Silverman, R. B., Radical ideas about monoamine-oxidase. Accounts Chem. Res. 1995, 28, (8), 335-342.
- Kalgutkar, A. S.; Castagnoli, N.; Testa, B., Selective inhibitors of monoamine-oxidase (MAO-A and MAO-B) as probes of its catalytic site and mechanism. Medicinal Research Reviews 1995, 15, (4), 325-388.
- Finberg, J. P. M.; Gillman, K., Selective inhibitors of monoamine oxidase type B and the "cheese effect". Int Rev Neurobiol 2011, 100, 169-190.
- Weyler, W.; Salach, J. I., Purification and properties of mitochondrial Monoamine-Oxidase type-A from human-placenta. Journal of Biological Chemistry 1985, 260, (24), 3199-3207.
- Newton-Vinson, P.; Hubalek, F.; Edmondson, D. E., High-level expression of human liver monoamine oxidase B in Pichia pastoris. Protein Expression and Purification 2000, 20, (2), 334-345.
- Li, M.; Hubalek, F.; Newton-Vinson, P.; Edmondson, D. E., High-level expression of human liver monoamine oxidase A in Pichia pastoris: Comparison with the enzyme expressed in Saccharomyces cerevisiae. Protein Expression and Purification 2002, 24, (1), 152-162.
- Albreht, A.; Vovk, I.; Mavri, J.; Marco-Contelles, J.; Ramsay, R. R., Evidence for a Cyanine Link Between Propargylamine Drugs and Monoamine Oxidase Clarifies the Inactivation Mechanism. Front. Chem. 2018, 6.
- Luhr, S.; Vilches-Herrera, M.; Fierro, A.; Ramsay, R. R.; Edmondson, D. E.; Reyes-Parada, M.; Cassels, B. K.; Iturriaga-Vasquez, P., 2-Arylthiomorpholine derivatives as potent and selective monoamine oxidase B inhibitors. Bioorganic & Medicinal Chemistry 2010, 18, (4), 1388-1395.
- Borstnar, R.; Repic, M.; Krzan, M.; Mavri, J.; Vianello, R., Irreversible inhibition of monoamine oxidase B by the antiparkinsonian medicines Rasagiline and Selegiline: A computational study. European Journal of Organic Chemistry 2011, 32, (32), 6419-6433.
- Repic, M.; Vianello, R.; Purg, M.; Duarte, F.; Bauer, P.; Kamerlin, S. C. L.; Mavri, J., Empirical valence bond simulations of the hydride transfer step in the monoamine oxidase B catalyzed metabolism of dopamine. Proteins-Structure Function and Bioinformatics 2014, 82, (12), 3347-3355.
- Basile, L.; Pappalardo, M.; Guccione, S.; Milardi, D.; Ramsay, R., Computational Comparison of Imidazoline Association with the 12 Binding Site in Human Monoamine Oxidases. Journal of Chemical Information and Modeling 2014, 54, (4), 1200-1207.
- Zapata-Torres, G.; Fierro, A.; Barriga-Gonzalez, G.; Salgado, J. C.; Celis-Barros, C., Revealing Monoamine Oxidase B Catalytic Mechanisms by Means of the Quantum Chemical Cluster Approach. Journal of Chemical Information and Modeling 2015, 55, (7), 1349-1360.
- Prah, A.; Purg, M.; Stare, J.; Vianello, R.; Mavri, J., How Monoamine Oxidase A Decomposes Serotonin: An Empirical Valence Bond Simulation of the Reactive Step. J. Phys. Chem. B 2020, 124, (38), 8259-8265.
- Mesiti, F.; Maruca, A.; Silva, V.; Rocca, R.; Fernandes, C.; Remiao, F.; Uriarte, E.; Alcaro, S.; Gaspar, A.; Borges, F., 4-Oxoquinolines and monoamine oxidase: When tautomerism matters. European Journal of Medicinal Chemistry 2021, 213.
- Edmondson, D. E.; Binda, C.; Mattevi, A., Structural insights into the mechanism of amine oxidation by monoamine oxidases A and B. Archives of Biochemistry and Biophysics 2007, 464, (2), 269-276.
- Binda, C.; Mattevi, A.; Edmondson, D. E., Structural properties of human monoamine oxidases A and B. International Review of Neurobiology 2011, 100, (100), 1-11.
- Edmondson, D. E.; Binda, C., Monoamine Oxidases. In Membrane Protein Complexes: Structure and Function, Harris, J. R.; Boekema, E. J., Eds. 2018; Vol. 87, pp 117-139.
- Edmondson, D. E.; Binda, C.; Wang, J.; Upadhyay, A. K.; Mattevi, A., Molecular and mechanistic properties of the membrane-bound mitochondrial monoamine oxidases. Biochemistry 2009, 48, (20), 4220-4230.
- Esteban, G.; Allan, J.; Samadi, A.; Mattevi, A.; Unzeta, M.; Marco-Contelles, J.; Binda, C.; Ramsay, R. R., Kinetic and structural analysis of the irreversible inhibition of human monoamine oxidases by ASS234, a multi-target compound designed for use in Alzheimer's disease. Biochimica Et Biophysica Acta-Proteins and Proteomics 2014, 1844, (6), 1104-1110.
- Sablin, S. O.; Krueger, M. J.; Singer, T. P.; Bachurin, S. O.; Khare, A. B.; Efange, S. M. N.; Tkachenko, S. E., Interaction of Tetrahydrostilbazoles with Monoamine-Oxidase-a and Monoamine-Oxidase-B. Journal of Medicinal Chemistry 1994, 37, (1), 151-157.
- Bocchinfuso, R.; Robinson, J. B., The stereoselectivity of inhibition of rat liver mitochondrial MAO-A and MAO-B by the enantiomers of 2-phenylpropylamine and their derivatives. European Journal of Medicinal Chemistry 1999, 34, (4), 293-300.
- Edmondson, D. E.; DeColibus, L.; Binda, C.; Li, M.; Mattevi, A., New insights into the structures and functions of human monoamine oxidases A and B. Journal of Neural Transmission 2007, 114, (6), 703-705.
- Lu, X.; Rodriguez, M.; Ji, H.; Silverman, R. B.; Vintém, A.-P. B.; Ramsay, R. R., Irreversible inactivation of mitochondrial monoamine oxidases. In Flavins and Flavoproteins 2002, Chapman, S. K.; Perham, R. N.; Scrutton, N. S., Eds. Rudolf Weber: Berlin, 2002; pp 817-830.
- Erdem, S. S.; Buyukmenekse, B., Computational investigation on the structure-activity relationship of the biradical mechanism for monoamine oxidase. Journal of Neural Transmission 2011, 118, (7), 1021-1029.
- Miller, J. R.; Edmondson, D. E., Structure-activity relationships in the oxidation of para- substituted benzylamine analogues by recombinant human liver monoamine oxidase A. Biochemistry 1999, 38, (41), 13670-13683.
- Orru, R.; Aldeco, M.; Edmondson, D. E., Do MAO A and MAO B utilize the same mechanism for the C–H bond cleavage step in catalysis? Evidence suggesting differing mechanisms. Journal of Neural Transmission 2013, Online First.
- MacMillar, S.; Edmondson, D. E.; Matsson, O., Nitrogen kinetic isotope effects for the Monoamine Oxidase B-catalyzed oxidation of benzylamine and (1,1-(2)H(2))benzylamine: nitrogen rehybridization and CH bond cleavage are not concerted. Journal of the American Chemical Society 2011, 133, (32), 12319-12321.
- Zenn, R. K.; Abad, E.; Kastner, J., Influence of the environment on the oxidative deamination of p-substituted benzylamines in monoamine oxidase. J. Phys. Chem. B 2015, 119, (9), 3678-3686.
- Umhau, S.; Pollegioni, L.; Molla, G.; Diederichs, K.; Welte, W.; Pilone, M. S.; Ghisla, S., The x-ray structure of D-amino acid oxidase at very high resolution identifies the chemical mechanism of flavin-dependent substrate dehydrogenation. Proceedings of the National Academy of Sciences of the United States of America 2000, 97, (23), 12463-12468.
- Akyuz, M. A.; Erdem, S. S., Computational modeling of the direct hydride transfer mechanism for the MAO catalyzed oxidation of phenethylamine and benzylamine: ONIOM (QM/QM) calculations. Journal of Neural Transmission 2013, 120, (6), 937-945.
- Oanca, G.; Stare, J.; Vianello, R.; Mavri, J., Multiscale simulation of monoamine oxidase catalyzed decomposition of phenylethylamine analogs. European Journal of Pharmacology 2017, 817, 46-50.
- Vianello, R.; Repic, M.; Mavri, J., How are Biogenic Amines Metabolized by Monoamine Oxidases? European Journal of Organic Chemistry 2012, (36), 7057-7065.
- Houslay, M. D.; Tipton, K. F., Reaction Pathway of Membrane-Bound Rat-Liver Mitochondrial Monoamine-Oxidase. Biochemical Journal 1973, 135, (4), 735-750.
- Houslay, M. D.; Tipton, K. F., Rat-Liver Mitochondrial Monoamine-Oxidase - Change in Reaction-Mechanism on Solubilization. Biochemical Journal 1975, 145, (2), 311-321.
- Pearce, L. B.; Roth, J. A., Human-Brain Monoamine-Oxidase Type-B - Mechanism of Deamination as Probed by Steady-State Methods. Biochemistry 1985, 24, (8), 1821-1826.
- Ramsay, R. R., Kinetic mechanism of monoamine oxidase-A. Biochemistry 1991, 30, (18), 4624-4629.
- Tan, A. K.; Ramsay, R. R., Substrate-specific enhancement of the oxidative half-reaction of Monoamine-Oxidase. Biochemistry 1993, 32, (9), 2137-2143.
- Edmondson, D. E.; Bhattacharyya, A. K.; Walker, M. C., Spectral and kinetic studies of imine product formation in the oxidation of p-(N,N-dimethylamino)benzylamine analogs by monoamine oxidase-B. Biochemistry 1993, 32, (19), 5196-5202.
- Woo, J. C. G.; Silverman, R. B., Monoamine oxidase B catalysis in low aqueous medium. Direct evidence for an imine product. Journal of the American Chemical Society 1995, 117, (5), 1663-1664.
- Edmondson, D., Hydrogen peroxide produced by mitochondrial monoamine oxidase catalysis: biological implications. Current pharmaceutical design 2014, 20 2, 155-60.
- Binda, C.; Hubalek, F.; Li, M.; Herzig, Y.; Sterling, J.; Edmondson, D. E.; Mattevi, A., Crystal structures of monoamine oxidase B in complex with four inhibitors of the N-propargylaminoindan class. Journal of Medicinal Chemistry 2004, 47, (7), 1767-1774.
- Dasgupta, S.; Mukherjee, S.; Mukhopadhyay, B. P.; Banerjee, A.; Mishra, D. K., Recognition dynamics of dopamine to human Monoamine oxidase B: role of Leu171/Gln206 and conserved water molecules in the active site cavity. Journal of Biomolecular Structure & Dynamics 2018, 36, (6), 1439-1462.
- Borštnar, R.; Repič, M.; Kamerlin, S. C. L.; Vianello, R.; Mavri, J., Computational Study of the pKa Values of Potential Catalytic Residues in the Active Site of Monoamine Oxidase B. J Chem Theory Comput 2012, 8, (10), 3864-3870.
- Ramsay, R.; Olivieri, A.; Holt, A., An improved approach to steady-state analysis of monoamine oxidases. Journal of Neural Transmission 2011, 118, (7), 1003-1019.
- Holt, A., On the practical aspects of characterising monoamine oxidase inhibition in vitro. Journal of Neural Transmission 2018, 125, (11), 1685-1705.
- Yu, P. H.; Bailey, B. A.; Durden, D. A.; Boulton, A. A., Stereospecific deuterium substitution at the alpha-carbon position of dopamine and its effect on oxidative deamination catalyzed by MAO-A and MAO-B from different tissues. Biochem Pharmacol 1986, 35, (6), 1027-36.
- Yu, P. H.; Davis, B. A., Inversion of selectivity of N-substituted propargylamine monoamine oxidase inhibitors following structural modifications to quaternary salts. International Journal of Biochemistry & Cell Biology 1999, 31, (12), 1391-1397.
- Geha, R. M.; Rebrin, I.; Chen, K.; Shih, J. C., Substrate and inhibitor specificities for human monoamine oxidase A and B are influenced by a single amino acid. Journal of Biological Chemistry 2001, 276, (13), 9877-9882.
- Milczek, E. M.; Binda, C.; Rovida, S.; Mattevi, A.; Edmondson, D. E., The 'gating' residues Ile199 and Tyr326 in human monoamine oxidase B function in substrate and inhibitor recognition. FEBS Journal 2011, 278, (24), 4860-4869.
- Hong, R. Y.; Li, X., Discovery of monoamine oxidase inhibitors by medicinal chemistry approaches. MedChemComm 2019, 10, (1), 10-25.
- Wu, S. M.; Qiu, X. Y.; Liu, S. J.; Sun, J., Single Heterocyclic Compounds as Monoamine Oxidase Inhibitors: From Past to Present. Mini-Reviews in Medicinal Chemistry 2020, 20, (10), 908-920.
- Rehuman, N. A.; Mathew, B.; Jat, R. K.; Nicolotti, O.; Kim, H., A Comprehensive Review of Monoamine Oxidase-A Inhibitors in their Syntheses and Poteneies. Combinatorial Chemistry & High Throughput Screening 2020, 23, (9), 898-914.
- Knez, D.; Colettis, N.; Iacovino, L. G.; Sova, M.; Pišlar, A.; Konc, J.; Lešnik, S.; Higgs, J.; Kamecki, F.; Mangialavori, I.; Dolšak, A.; Žakelj, S.; Trontelj, J.; Kos, J.; Binda, C.; Marder, M.; Gobec, S., Stereoselective Activity of 1-Propargyl-4-styrylpiperidine-like Analogues That Can Discriminate between Monoamine Oxidase Isoforms A and B. Journal of Medicinal Chemistry 2020, 63, (3), 1361-1387.
- Orayj, K.; Lane, E., Patterns and Determinants of Prescribing for Parkinson’s Disease: A Systematic Literature Review. Parkinson’s Disease 2019, 2019, 9237181.
- Binde, C. D.; Tvete, I. F.; Gasemyr, J.; Natvig, B.; Klemp, M., A multiple treatment comparison meta-analysis of monoamine oxidase type B inhibitors for Parkinson's disease. British Journal of Clinical Pharmacology 2018, 84, (9), 1917-1927.
- Fowler, J. S.; Logan, J.; Shumay, E.; Alia-Klein, N.; Wang, G.-J.; Volkow, N. D., Monoamine oxidase: radiotracer chemistry and human studies. Journal of Labelled Compounds & Radiopharmaceuticals 2015, 58, (3), 51-64.