Metal/lipase-combo catalyzed dynamic kinetic resolution (DKR) of racemic chiral alcohols is a general and practical process to obtain the corresponding enantiopure esters R with quantitative conversion. The use of known Ru-catalysts as well as newly developed homogeneous and heterogeneous metal catalysts (Fe, V) contributed to make the DKR process more sustainable and to expand the substrate scope of the reaction. In addition to classical substrates, challenging allylic alcohols, tertiary alcohols, C1-and C2-symmetric biaryl diols turned out to be competent substrates. Synthetic utility further emerged from the integration of this methodology into cascade reactions leading to linear/cyclic chiral molecules with high ee through the formation of multiple bonds, in a one-pot procedure.
Note: The entry will be online only after author check and submit it.
1. Introduction
Two enantiomers of a chemical compound may have different biological activity resulting from the interaction with chiral environment in biological systems. Thus, it is highly desirable to have easy access to enantiomerically pure molecules which find application as pharmaceuticals, agrochemicals, flavors and fragrances. Many of them originate from alcohols and a lot of work has been done to develop efficient methods for their synthesis in enantioenriched form
[1].
Catalytic asymmetric synthesis is based on the enantioselective construction of a new stereocenter by using a chiral catalyst. The biocatalytic reduction of prochiral ketones in the presence of ketoreductases is an example of the enzymatic approach while the hydrogenation reaction of the same substrates catalyzed by chiral transition-metal complexes well represents the chemical strategy
[2][3][2,3]. Other asymmetric transformations utilize racemates which are often easier and cost-effective to synthesize. In this case, the transformations can proceed through kinetic resolution promoted by an artificial chiral catalyst or an enzyme
[4][5][4,5]. In industry the predominant method to access enantiomerically pure products is still the kinetic resolution (KR) of racemates promoted by enzymes
[4][5][4,5]. Lipases are widely used in the KR of racemic alcohols: they catalyze either hydrolysis reaction or ester synthesis even in the presence of organic solvents
[5][6][5,6]. The chiral enzyme preferentially reacts with the
R-enantiomer of a racemic alcohol which can be transformed at a higher rate (k
R >> k
S) (
Scheme 1a). Kinetic resolution proceeds with high enantioselectivity when the difference in the enantiomer reaction rates, namely the enantiomeric ratio E (k
R/k
S), is ≥100.
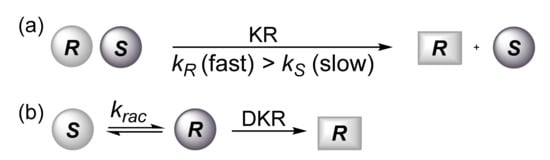
Scheme 1. (a) Kinetic resolution. (b) Dynamic kinetic resolution.
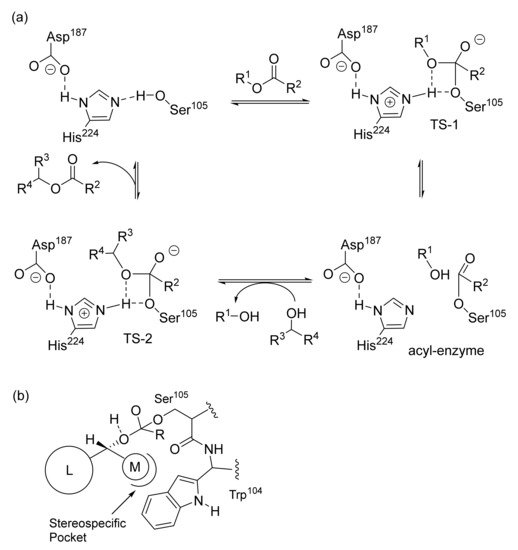
Lipase enantioselectivity can be rationalized considering their active site which is formed by a “catalytic triad” including residues of a serine, a histidine and an aspartate (
Scheme 2a)
[5][6][5,6]. The enantioselective esterification of a racemic secondary alcohol occurs in the presence of an ester acting as an acyl donor. The latter interacts with the OH serine residue made more nucleophilic by H-bonding interactions. Then, the resulting acyl-enzyme intermediate preferentially reacts with the (
R)-enantiomer which fits better in the active site (
Scheme 2b). The substituents bonded to the stereocenter of the substrate are placed in two different pockets depending on their size: the medium-sized substituent (M) is located inside the stereospecificity pocket, while the large one (L) points towards the entrance of the active site
[7][8][9][7,8,9]. The transesterification reaction leads to the corresponding (
R)-ester and free enzyme, ready for a new cycle.
Scheme 2. (a) Mechanism of alcohol acylation catalyzed by lipase. (b) Favored (R)-enantiomer. M stands for a medium substituent; L stands for a large substituent.
The (
R)-ester can be obtained with a maximum yield of 50%. This limitation can be overcome when the lipase-catalyzed transesterification is combined with the fast and continuous in situ interconversion of the less reactive (
S)-enantiomer into the more reactive one (
Scheme 1b). Racemization is normally an undesired reaction in asymmetric synthesis, but its combination with a resolution process may become a powerful method called dynamic kinetic resolution (DKR). The latter is effective when the rate of racemization of the unwanted (
S)-enantiomer is faster than the rate of transesterification (k
rac > k
S). In this case, the (
R)-ester can be obtained with a theoretically 100% yield (
Scheme 1b).
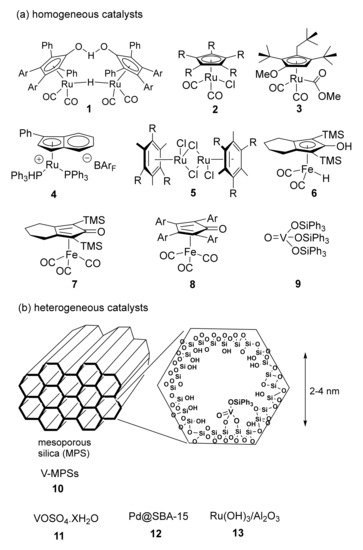
Different racemization methods employing bases, acids or enzymes as catalysts have been developed in accordance with the type of substrate used. A key-point for the success of the DKR is the compatibility between the two catalysts and the conditions (temperature and pH) required for both the kinetic resolution of the racemate and the catalytic enantiomer racemization. Alcohols easily undergo racemization in the presence of metal catalysts (Al, Ir, Ru, Rh, Pd). After the pioneering works of Williams and Bäckvall
[10][11][10,11], a number of homogeneous and heterogeneous transition metal-based catalysts
[12][13][12,13] have been developed and employed as co-catalysts in the DKR of a variety of racemic chiral alcohols (
Scheme 3)
[14][15][16][17][14,15,16,17].
Scheme 3. Selected examples of effective catalysts (homogeneous and heterogeneous) for alcohol racemization. ˉBArF = tetrakis[3,5-bis(trifluoromethyl)phenyl]borate.
In addition to the initially used enzymes, various synthetic metal complexes were recently applied as single catalysts to promote dynamic kinetic processes
[18]. For instance, chiral palladium complexes were reported to catalyze both the KR of a racemic substrate and the rapid racemization of the slow reacting enantiomer leading to the same enantiomerically enriched alcohol derivative
[19]. It is important to mention that the desymmetrization of racemates can also occur through different paths including dynamic kinetic asymmetric transformations (DYKATs) and direct enantio-convergent transformations (DETs). A DYKAT proceeds through the irreversible symmetrization of the racemic substrate leading to a prochiral intermediate
[20]. Finally, a DET of a racemate into a single chiral alcohol or its derivative implies that each of the two enantiomers reacts through different pathways
[21][22][21,22].
2. DKR of Alcohols Co-Catalyzed by Homogeneous Catalysts (Ru, Fe)
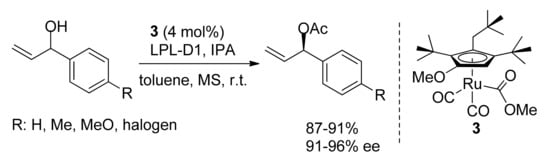
The development and application of new redox catalysts may improve the scope of the reaction. In 2019, Park and Kim reported that Ru-Complex
3 enabled the selective racemization of several secondary alcohols including α-substituted allylic alcohols. They were quantitatively transformed into the corresponding esters in the presence of LPL-D1, a lipoprotein lipase from
Burkholderia species (
Scheme 4)
[23]. It is to note that the DKR of α-substituted allylic alcohols co-catalyzed by ruthenium redox-catalysts may be less selective as the substrates can undergo a competitive metal-catalyzed double bond isomerization leading to the corresponding saturated ketones (see
Section 5)
[24][25][24,25]. Apparently, this side-reaction is favored in the presence of a base usually used as an additive
[12][13][12,13]. Interestingly, the unique feature of Precatalyst
3 is that its employment does not require any base (see
Section 5).
Scheme 4. DKR of α-arylallyl alcohols. LPL-D1: anionic surfactant-treated lipoprotein lipase. IPA: isopropenyl acetate. MS: molecular sieves.
Sustainable metal catalysis relies on the use of inexpensive, abundant and non-toxic metals. Among them, iron is the most abundant in earth’s upper crust
[26]. Most of the examples of DKR are co-catalyzed by homogeneous metal complexes containing precious metals. In the last ten years, the development of homogeneous catalysts based on biorelevant metals capable of promoting a variety of reactions including oxidation and reduction has become a hot topic
[27]. However, their integration in a DKR process may be demanding because of the incompatibility with the biocatalytic system
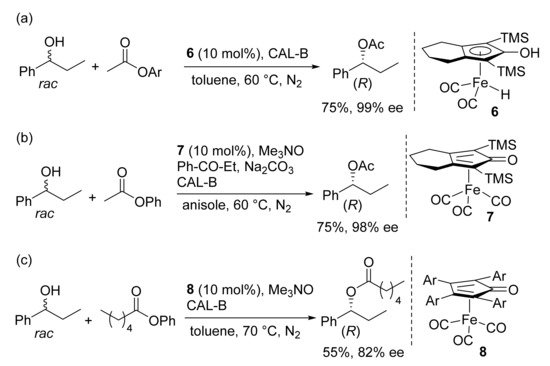
[28]. In 2016, Rueping and coworkers found that iron Complex
6 is a competent co-catalyst in the DKR of racemic secondary alcohols using commercially available immobilized lipase B from
Candida antarctica CAL-B (Novozym
® 435) and
p-chlorophenylacetate, as the acyl donor (14 examples, 68–95% yield, 92–99% ee) (
Scheme 5a)
[29]. It is to note that in comparison to precious metal catalysts, iron complexes turned out to be less reactive and the corresponding co-catalyzed DKR occurred at higher temperature (60 °C). Catalyst
6, which is sensitive to air and moisture, was replaced with the stable and easy to handle Complex
7 by Bäckvall, in 2017 (
Scheme 5b)
[30]. The latter is a precatalyst and can be activated in situ through an oxidative partial decarbonylation of the iron centre promoted by Me
3NO (
Scheme 5). An inorganic base and a catalytic amount of the ketone intermediate were also needed to make the racemization reaction more efficient. Racemic 1-arylethanols bearing
p-halogen, CF
3 and MeO substituents were quantitatively transformed into the corresponding enantioenriched acetates in fair to good selectivity (nine examples, 62–83% yield, 95–99% ee). Interestingly, no additives were required when tricarbonyl(cyclopentadienone)iron Complexes
8 (Ar: Ph, 4-MeOC
6H
4) were employed (
Scheme 5c)
[31]. Moderate to high conversions of a variety of secondary aliphatic and benzyl alcohols were obtained, under DKR conditions (18 examples, esters
R: 42–94% yield, 82–99% ee).
Scheme 5. Iron-catalyzed DKR of chiral secondary alcohols. TMS: trimethylsilyl group. Ar: 4-MeOC6H4.
Kinetic resolution is highly selective with centrochiral molecules as lipases, typically CAL-B, can differentiate the geometry of tetrahedral carbon centers. Aryl or heteroaryl alkyl (Me, Et) carbinols, aryl-substituted propargylic alcohols, 1,2-diarylethanols and benzoins are substrates that give excellent results in terms of selectivity when subjected to the DKR process
[14][15][16][17][14,15,16,17]. The reaction is less effective when secondary aryl carbinols contain alkyl groups which are branched in the α, β, or γ position, as they do not fit well in the medium-sized pocket. The DKR of simple secondary aliphatic alcohols as well as those bearing additional functional groups (CN, Cl, N
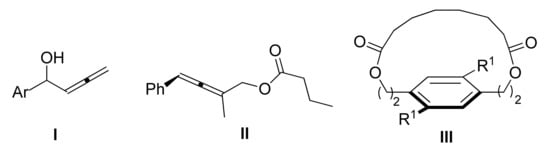
3, ester, phosphate, amides, ether) proceeds with moderate to excellent selectivities, in some cases being sensitive to the length of the alkyl chain or to the presence of a branched alkyl group. Among centrochiral molecules, α-allenic alcohols of Type I still represent an unsolved problem in chemoenzymatic resolution (
Scheme 6). Lipase IL1-PS (IL1-supported lipase PS) could promote selective KR, but none of the commonly used Ru-complexes turned out to be an effective racemization catalyst for such compounds
[32].
Scheme 6. Challenging substrates for dynamic kinetic resolution process.
The potential of lipases in the synthesis of chiral esters is not limited to C-centrochiral alcohols. Planar chiral macrocycle III (R
1: Br, 80%, 99% ee) was recently synthesized through an atroposelective macrocyclization of prochiral primary diol with a diester in the presence of CAL-B as a catalyst
[33]. Nevertheless, axially planar or helically chiral alcohols have been less exploited as potential substrates for DKR. A rare example is the chemoenzymatic synthesis of the (
R)-ester of axially chiral primary allenic alcohol II with vinyl butyrate, using a dimeric
N-heterocyclic carbene palladium complex and porcine pancreatic lipase, as co-catalysts (83% yield and 89% ee)
[34].
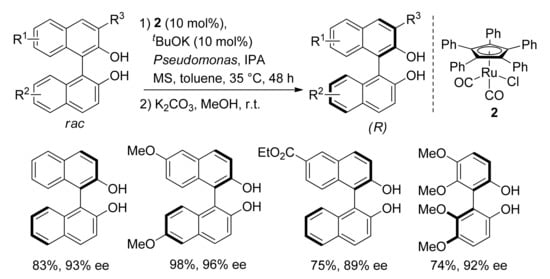
In 2018, Akai and coworkers reported the first synthesis of axially chiral 2,2′-dihydroxy-1,1′-biaryls through the DKR of the corresponding racemates followed by the hydrolysis of the ester intermediates (
Scheme 7)
[35]. Co-catalysis of the immobilized
Pseudomonas sp. lipoprotein lipase and Complex
2 (R = Ph) led to the optically active biphenol or binaphthol derivatives in fair to excellent yields and enantioselectivity (16 examples, 61–98% yield, 86–98% ee). Lower yields were obtained in the presence of electron-deficient substituents (Br, ester and alkyl groups). Apparently, the racemization occurs through a redox mechanism involving a radical intermediate which represents a novelty in the case of Catalyst
2 (see
Section 5). Optically active C
1- and C
2-symmetric atropoisomeric biphenol or binaphthol derivatives are used as building blocks for natural product and chiral ligand synthesis. This approach is a good alternative to conventional procedures based on the metal-catalyzed enantioselective oxidative coupling of 2-naphthol or biphenol derivatives
[36].
Scheme 7. DKR of 2,2-dihydroxy-1,1′-biaryls: selected examples.
3. DKR of Alcohols Co-Catalyzed by Heterogeneous Catalysts (V, Pd)
Almost any type of catalytic species can be immobilized on heterogeneous supports which should be: (a) inert to avoid unwanted side reactions decreasing the yield of the desired product, (b) highly stable over a wide range of reaction conditions to enable the re-use of the catalyst
[17]. Then, the development of DKR protocols with efficient and reusable catalysts would be highly desirable. Heterogeneous vanadium catalysts are still playing a central role in the chemoenzymatic resolution of alcohols (
Scheme 3)
[37]. Vanadium is a biorelevant metal, being abundant in nature and relatively low toxic. In 2006, Akai and Kita began to use homogeneous oxovanadium species
9 as a racemization catalyst (see
Section 5) in combination with Novozym
® 435
[38]. However, improved racemization activity was obtained when the homogeneous species was covalently attached to the inner surface of a neutral solid carrier such as mesoporous silica (MPS)
[39]. The resulting heterogeneous Catalyst
10 was employed in the DKR of various chiral secondary alcohols including allylic alcohols
[40].
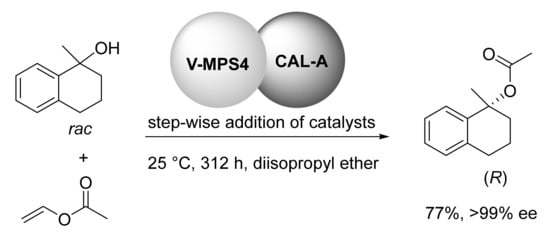
The development of enantioselective synthesis of tertiary alcohols is a challenge in organic synthesis
[41]. The application of the DKR method relies on the availability of an efficient racemization method and its compatibility with the enzyme. As the racemization of tertiary alcohols cannot be achieved through a catalytic redox process, an alternative way could be based on the cleavage of the C-O bond leading to a carbocation as an intermediate (see
Section 5). Recently, tertiary alcohols have been reported to undergo complete and selective racemization in the presence of an acidic resin material, but no data are available about the combination with a lipase
[42]. Remarkably, in 2020 Akai and Gröger found that V-MPS triggered the racemization of (
S)-1,2,3,4-tetrahydronaphthalene-1-ol (25 °C, 24 h, 44%→16% ee)
[43]. The co-catalyzed DKR of the tertiary alcohol in the presence of CAL-A from
Candida antartica lipase A and vinyl acetate led to the desired (
R)-acetate in 77% yield and excellent ee value (>99%) (
Scheme 8). Stepwise addition of both the catalysts during the reaction allowed to overcome the inactivation of the enzyme due to long reaction times (312 h).
Scheme 8. DKR of a tertiary alcohol.
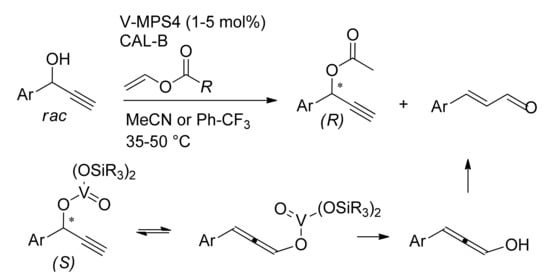
The DKR of propargylic alcohols carried out in (trifluoromethyl)benzene or MeCN as solvents, led to the corresponding (
R)-esters with high selectivity (15 examples, 70–99%, 81–99% ee) (
Scheme 9)
[44]. The choice of the solvent was crucial to reduce or totally suppress the formation of the unsaturated aldehyde which can form as a by-product. Propargyl vanadate (
S) which is involved in the racemization of the alcohol could transform into the allenyl-vanadate intermediate through Meyer–Schuster rearrangement (
Scheme 9). Subsequent hydrolysis and the tautomerization of the resulting alcohol led to the undesired aldehyde.
Scheme 9. DKR of propargylic alcohols: competition between racemization and rearrangement.
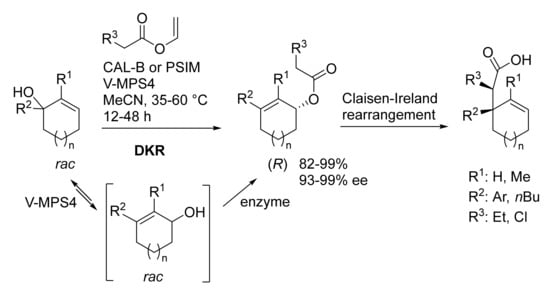
Tertiary cyclohexenols were selectively converted into the corresponding secondary allylic esters (
R) through dynamic kinetic resolution and 1,3-allylic transposition promoted by Lipase/V-MPS4 as co-catalysts (
Scheme 10)
[45]. The DKR is the first step of an interesting enantio- and chemo-selective reaction sequence which continues with a Claisen–Ireland rearrangement leading to optically active cycloalkenes bearing all-carbon quaternary centers. They are interesting building blocks used for the synthesis of natural compounds. Reusability of the immobilized lipases and vanadium catalyst made the reaction more efficient and attractive.
Scheme 10. Dynamic kinetic resolution as a key step in the synthesis of optically active cycloalkenes. PSIM: immobilized Burkholderia cepacia lipase.
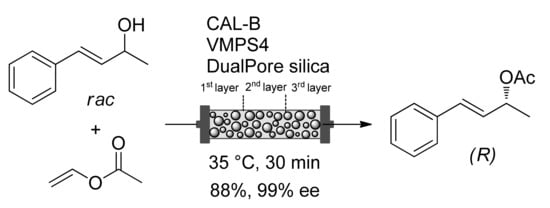
Heterogeneous supported catalysts find application in flow reactors for continuously running processes. The use of packed-bed reactors improves the turnover numbers and lifetimes of the catalysts by avoiding their recovery for reuse
[46]. They present advantages including better mass and heat transfer, improved safety profiles, easier work-up and accurate control of reaction parameters (temperature, pressure, time). Continuous-flow processes have higher productivity and reproducibility than batch reactions
[47]. In 2020, the groups of Gröger and Akai developed the continuous-flow DKR of allylic alcohols. A three-layer packed column containing DualPore silica beads as a filler, and both catalysts with increasing ratio of V-MPS4/CAL-B was used (
Scheme 11)
[48]. Alcohols were converted after 30–60 min of residence time and the corresponding (
R)-esters were isolated in 88–92% yield and 96–99% ee. Noteworthy, the flow DKR of an aromatic allylic alcohol continuously conducted for three days turned out to be highly selective using a lower number of catalysts in comparison to that of batch reaction.
Scheme 11. Continuous-flow DKR of an allylic alcohol.
A drawback of V-MPS catalyst is the cost of (a) the mesoporous silica matrix used to support the homogeneous vanadium oxide, (b) the procedure for immobilization. In 2020, Milagre and coworkers applied the less expensive hydrated vanadyl sulfate (VOSO
4·XH
2O) in combination with Novozym
® 435 for the DKR of various benzylic alcohols with vinyl decanoate at 50 °C, in heptanes (10 examples, 74–91%, up to 99% ee)
[49]. Both the heterogeneous chemo- and biocatalysts could be re-used up to five times without a significant decreasing of the conversion. The catalytic system worked well with benzyl alcohols bearing methyl/ethyl side-chains as they react faster (8 h) than alcohols bearing larger groups. Unfortunately, prolonged reaction time caused catalyst incompatibility thus precluding the success of the reaction.
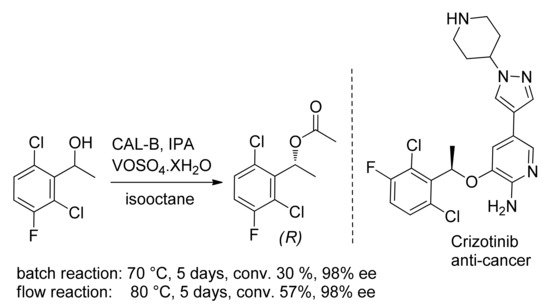
A chiral benzyl alcohol, namely the (
R)-1-(2,6-dichloro-3-fluorophenyl)ethanol is the key-intermediate in the synthesis of Crizotinib (
Scheme 12). The continuous flow-DKR of the racemate using Novozym
® 435 and VOSO
4·XH
2O allowed to circumvent the incompatibility between bio- and chemo-catalyst: better conversion was obtained in comparison to batch reaction
[50].
Scheme 12. DKR of 1-(2,6-dichloro-3-fluorophenyl)ethanol, a key-precursor of Crizotinib.
A heterogeneous catalyst based on size-controlled (2.8 nm) palladium nanoparticles highly dispersed on mesoporous silica, as a support (Pd@SBA-15) was synthesized in 2017 by Li and coworkers
[51]. The catalyst promoted the complete racemization of (
S)-1-phenylethanol in hexane at 70 °C, under H
2 pressure (0.03 MPa) and microwave irradiation. The latter was crucial to speed up the racemization on the Pd surface, while an appropriate H
2 pressure was needed to prevent the formation of the ketone intermediate (catalytic alcohol oxidation). The co-catalyzed DKR of chiral benzyl alcohols with CAL-B and vinyl acetate worked well independently on the electronic nature of the aromatic substituents (H, Me, Cl, MeO) (2–4 h, up to 89% yield and 99% ee). Both catalysts were easily separated and re-used six times without loss of efficiency. In comparison to zeolites or Ru(OH)
3 as co-catalysts, the use of
12 gave similar yield and better enantioselectivity
[17].