Sphingolipids (SLs) are highly abundant components of cellular membranes and as such, are essentially involved in their biophysical and signaling properties. A complex metabolic network consisting of enzymes catalyzing their synthesis, modification (phosphorylation, glycosylation) and breakdown regulates accumulation of sphingolipid species and thereby the sphingolipid pool at rheostat conditions, and this can undergo substantial changes in response to metabolic and external challenges. This has been excellently reviewedand will therefore just be briefly re-iterated below.
- glycosphingolipids
- ceramides
- sphingosine 1-phosphate
- sphingomyelinase
- HIV
- SARS-CoV-2
- measles
1. Introduction
Sphingolipids (SLs) are highly abundant components of cellular membranes and as such, are essentially involved in their biophysical and signaling properties. A complex metabolic network consisting of enzymes catalyzing their synthesis, modification (phosphorylation, glycosylation) and breakdown regulates accumulation of sphingolipid species and thereby the sphingolipid pool at rheostat conditions, and this can undergo substantial changes in response to metabolic and external challenges. This has been excellently reviewed [1,2,3][1][2][3] and will therefore just be briefly re-iterated below. Depending on the length of their fatty acid chains and their degree of saturation, SL species have a strong impact on biophysical membrane parameters such as fluidity or rigidity and curvature, and on interaction with membrane proteins and/or cytoskeletal components, and membrane compartmentalization. This coins their ability to promote the formation and activity of signaling platforms in a dynamic and spatiotemporally regulated manner [4,5,6,7][4][5][6][7]. In addition to structurally supporting the interaction of receptors with their membrane proximal signalosome components, certain SL species (especially ceramides, ceramide-1-phosphate, sphingosine and sphingosine 1-phosphate) act as highly potent signaling molecules themselves, regulating, for instance, cellular apoptosis and autophagy, or activation and survival, respectively [1,2,8,9,10,11,12][1][2][8][9][10][11][12]. Therefore, dynamic alterations of the SL pool and its subcellular compartmentalization not surprisingly have a substantial impact on most cellular processes determining viability and responsiveness. Supporting their decisive role also in clinical terms, alterations in SL biosynthesis or accumulation are of crucial importance in the pathophysiology in severe diseases including lysosomal storage diseases and cancer, and pharmacological interference with SL metabolism has already been proven as an effective target for treatment of major depression, cancer and inflammation [10,13,14,15,16][10][13][14][15][16].
Lack of suitable reagents for detection and fixation or protocols for quantification of SL species have long hampered detailed studies in SL biology at a cellular level, while on an organismic level, ubiquitous genetic ablation of SL modifying enzymes in mice, in some cases reliably reproducing disease processes in humans, precluded detailed analysis on the pathophysiological role of individual compartments. With the advent of bio-orthogonally functionalized SLs, important progress has been made in their detection and recording their trafficking at high resolution, identifying associated protein complexes, and, in combination with compartment-specific targeting of SL modifying enzymes, determining topology and biological consequences of their metabolism [17,18,19,20,21,22][17][18][19][20][21][22]. Mass spectrometry is the favored analytical technology for SL analysis. Thus, this technique has the capability to both acquire sensitive and quantitative measurements and to unravel the molecular intricacies of SL species. At present, mass spectrometric analysis of lipid extracts, mainly by infusion electrospray ionization, is utilized to determine sensitive and quantitative information of specific SLs in biological processes. As a complementary method, mass spectrometry imaging, albeit less quantitative and less specific, provides information of spatial SL distribution in tissues.
These techniques are being exploited in the context of general SL biosynthesis and turnover, which, due to its central role in cell physiology, directly translates into regulation of cellular processes, and therefore has been targeted for therapeutic intervention as referred to above. The last two decades have seen an increasing amount of studies addressing the role of the SL pathway in infections where their turnover has been found to regulate the disease process both at the level of the pathogens’ life cycle and efficiency of immune control. Rather than discussing that process in bacterial infections (which has been excellently reviewed [23,24,25,26][23][24][25][26]), this review focuses on the role of sphingolipid metabolites in viral infections. Especially the importance of the SLs in governing essential steps in virus–host cell interaction is described.
2. Sphingolipid Metabolism
Sphingolipid de novo synthesis is initiated in endoplasmic reticulum (ER) by serine palmitoyl-transferase (SPT) catalyzed condensation of the activated C 16 fatty acid palmitoyl-CoA and the amino acid l -serine to yield 3-ketosphinganine. This is then rapidly reduced to sphinganine by 3-ketosphinganine reductase (3KSR) in a NADPH-dependent manner. Then, sphinganine is further N -acylated by the action of six ceramide synthase isoforms (CerS1-6) encoded by six distinct genes to form ceramides [1]. The most notable characteristic of the individual CerS isoforms is a different acyl-CoA preference that can overlap within their isoforms. Variations of acyl-chain length in a cell type specific manner result in ceramide species with different biophysical properties and distinct biological functions. It has recently been possible to fully reconstitute and to monitor this de novo ceramide synthesis pathway in vitro by using both stably isotope labeled or bio-orthogonally modified precursors ( Figure 1 ) [27,28][27][28].
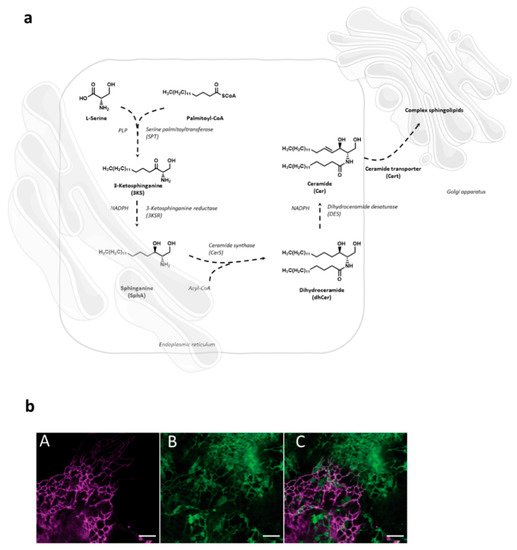
Following transfer to the Golgi compartment, ceramides are further modified by glycosylation to yield glucosylceramides or acquisition of a phosphocholine headgroup via sphingomyelin synthase to yield sphingomyelin, both of which are transported to the plasma membrane using vesicular exocytosis where they, according to the topology of the modifying enzymes to the Golgi luminal compartment, predominantly localize to the outer membrane leaflet. As for other membrane lipids, trans-bilayer asymmetric distribution of SLs in the plasma membrane is regulated by the activity of scramblases at the steady state, or actively, by ATP-dependent flippases or floppases, and this is crucial for membrane integrity, charge and compartmentalization (reviewed in [29]). SL catabolism is initiated by ceramide production through the activity of sphingomyelinases or in the case of glycosphingolipids (GSLs) via specific hydrolases. Sphingomyelinases, depending on their pH optima, are grouped into neutral, acid and alkaline isoforms. Best studied amongst those include the neutral sphingomyelinase 2 (NSM2) which resides in association with the cytosolic membrane leaflet of the plasma membrane, multi-vesicular body, or the Golgi compartment, and is activated in response to a variety of signals including stress, cytokines or T cell receptor ligation [30,31,32,33][30][31][32][33]. Equally well investigated, acid sphingomyelinase 1 (ASM) anchors to anti-cytosolic membrane leaflets in the late endosomal compartment from where it is either secreted (soluble, sASM) or displayed after fusion with the plasma membrane in response to receptor signaling or during exocytic membrane repair [13,34][13][34]. Ceramide levels are tightly controlled as is reflected by its rapid conversion into ceramide 1-phosphate or de-acylation by ceramidases into sphingosine which, accumulating to even lower levels, serves as target for phosphorylation by sphingosine kinases to yield sphingosine 1-phosphate (S1P), a potent signaling molecule. Reflecting the dynamic demands to the system, anabolic and catabolic steps in SL metabolism are reversible except for the final breakdown of S1P into phospho-ethanolamine and hexadecenal by S1P lyase which thereby terminates the SL life cycle (reviewed in [2,35][2][35]). Notably, its phosphorylation renders S1P sufficiently polar and thereby soluble in the cytosol, from where it is also exported to initiate autocrine and paracrine S1P receptor signaling ( Figure 2 ) [36,37][36][37].
3. Sphingolipid Targets in Viral Life Cycles
Firm attachment to and passage of the host cell plasma membrane effectively initiate and thereby enable viral replication and therefore, the role of SLs in these processes has most intensely been studied. In particular, the role of membrane microdomains enriched for particular SL species and the impact of their accumulation or metabolization there has been studied with regard to receptor segregation, membrane biophysical alterations (favoring fusion or endocytosis) as well as initiation of signaling cascades involved in uptake and trafficking.
When evaluated at an overall level, individual SL species accumulating in viral host cells may be favorable for either the host cell or the virus. Lipidomic profiling in a bronchial epithelial cell line revealed massive perturbation of particularly the SL pathway, and therein, most prominently in sphingomyelin species, after infection by influenza A virus, rhinovirus and SARS-CoV-2. Revealing that requirements of sphingomyelin in viral replication might substantially differ, exposure of infected cells to bacterial sphingomyelinase suppressed replication of influenza virus and SARS-CoV-2, yet enhanced that of rhinovirus [109][38]. Additionally, in lung epithelial cells, a protective role of ceramides was evidenced where replicating, but not UV-inactivated IAV caused de novo biosynthesis of ceramide which limited viral replication [110][39] as previously also suggested for hepatitis B [111][40]. In contrast, SLs, also including ceramides, may also act pro-virally by supporting viral replication as revealed for hepatitis C, West Nile and Dengue viruses [112,113,114][41][42][43].
The membrane patch where viral assembly occurs defines the composition of the viral particle’s envelope membrane. Initially established as highly relevant in membrane model systems, the operation of lipid-based protein sorting in mammalian cell membranes has in fact been pioneered by studies on HIV biogenesis [134,135][44][45]. When comparatively analyzed with that of the overall cellular membrane, the HIV particle substantially differs in its lipid composition as especially reflected by selective enrichment of SM and dihydro-sphingomyelin, while ceramides are barely represented [136,137][46][47]. This suggested that the viral core either selects already existing or actively remodels host cell membranes during the assembly and subsequent budding process. It was only after techniques to visualize and trace single virus assembly by quantitative live cell imaging that, driven by oligomerized HIV Gag protein at the inner membrane leaflet, formation of ordered membrane domains orchestrating lipid and protein composition could be demonstrated [135,138][45][48]. Underlying mechanisms described in this and earlier highly elegant studies included acquisition of membrane curvature [139[49][50],140], lipid-based phase partitioning and sequential sorting of proteins, and supported the importance of trans-bilayer coupling by acyl chain interactions for phase separation of the outer membrane leaflet assembly site.
Finally, the lipid composition of the enveloped viral particle may substantially impact its infectivity. Suggesting that SM content of the particle is important, viral entry was reduced after treatment of bovine herpesvirus particles, but not that of the target cells with bacterial sphingomyelinase (bSMase). In contrast, pseudorabies virus entry was sensitive to bSMAse at the level of the host cell and not the particle, while HSV-1 entry was insensitive to bSMase exposure of either membrane or particle [144][51] and SM was found to be important in IAV infection both at the viral particle and the host cell level [145][52]. As especially revealed for HIV and Ebola virus particles, viral uptake into DCs is substantially enhanced upon recognition of sialylated gangliosides anchored to viral membranes by Siglec-1 [146,147][53][54].
4. Outlook and Perspectives
Drug repurposing has already revealed the potential but also limitations of SL pathway modulating compounds in containing viral infections. This has so far been studied for the SphK/S1P/S1P lyase system and results obtained clearly document the complexity of responses. FTY720 (commercially known as fingolimod or Gilenya), upon phosphorylation by SphK2, acts to inhibit S1P receptor signaling, and thereby, potently inflammation and is therefore licensed for treatment of multiple sclerosis. A recent study suggested fingolimod as a promising novel therapy approach for HIV treatment and prevention. Firstly, it prevented viral spread in human CD4+ T cells by reducing surface density of this receptor (which may be of particular relevance given the low density of HIV glycoproteins available for interaction), and secondly, it enhanced the activity of SAMHD1, a cellular restriction factor, and thereby, levels of total and integrated HIV [150][55]. Thus, in addition to preventing S1P-mediated cellular activation which overall is mainly beneficial for viral replication, fingolimod has apparently virus-specific targets as well. Although it did not mechanistically address the role of S1P or S1PR signaling, a recent study provided compelling evidence that SphK2, the enzyme required to activate fingolimod, efficiently prevented clearance of LCMV in experimentally infected mice by restricting T cell immunopathology and promoting viral persistence [151][56]. Based on its anti-inflammatory activity, fingolimod (or, in general terms, inhibitors of S1P receptor signaling) has also been evaluated for its beneficial effect in virally-induced immunopathology when brought about by hyperinflammation through the activity of immune effector cells or cytokine storm. Thus, S1P analogs effectively reduced lung pathology induced by experimental influenza or paramyxovirus infection in mice [152,153,154][57][58][59]. Cytokine storm and hyperinflammation associated with prominent lung infiltration of CD8+ T cells and NK cells also marks the late phase of SARS-CoV-2 infection in mice, and therefore, S1P analogs in COVID-19 therapy might be an option [122][60]. This needs, however, to be critically assessed because interference with S1PR signaling obviously also limits the ability of the host to clear viral infection by both disabling recruitment and activation of effector cells and by limiting IFN-responses [152,155][57][61]. Interestingly, a host-protective role has been revealed for the S1P lyase in vitro; independently of its ability to catalyze S1P this enzyme enhanced type I interferon production in influenza virus infection [156][62] .
Another example where targeted intervention of the SL pathway may be highly effective in viral pathogenesis is production of extracellular vesicles (EV). These come in different types, dependent on their origin, and production of least some EV species was found to occur clearly ASM or NSM-dependently [157,158,159,160][63][64][65][66]. Viral infections are known to affect generation and content of EVs, and thereby mediate transfer of both viral and host cell proteins or nucleic acids [161][67]. Viral components transferred by this pathway include single proteins, sub-genomic or micro-RNAs, up to entire viral particles, the latter revealed for hepatitis C and A viruses. Alternatively, EVs transfer cellular or viral proteins involved in regulating the interferon response of yet uninfected neighboring cells. For instance, EVs were found to amplify the innate immunity to hepatitis B, mouse hepatitis and adenovirus in liver cells in vitro and in vivo by transfer of interferon effector proteins from infected cells to uninfected cells, and importantly, this was prevented by both pharmacologic inhibition and genetic ablation of NSM2 (in vivo mediated by hydrodynamic injection of siRNA) [162][68]. Targeting the activity of SL metabolizing enzymes involved in EV production might thus be highly efficient in regulating viral pathogenesis at multiple levels.
Apparently, yet not surprisingly, the impact of the SL anabolic and catabolic pathway on viral replication occurs at multiple levels, and doubtlessly, much more will be detected in the future now that experimental systems have advanced. This especially refers to mouse models allowing for—ideally conditional—genetic ablation of enzymes involved in the SL pathway, in particular compartments that will be of particular relevance to study its role in viral pathogenesis and to evaluate the potential of pharmacologic intervention. It is, however, essentially clear that interventions into viral replication per se are highly effective when implemented early in infection, and thereby need to be complemented by drugs modulating induction and activity of host immune responses.
References
- Hannun, Y.A.; Obeid, L.M. Many Ceramides. J. Biol. Chem. 2011, 286, 27855–27862.
- Hannun, Y.A.; Obeid, L. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150.
- Harayama, T.; Riezman, H. Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296.
- Bieberich, E. Sphingolipids and lipid rafts: Novel concepts and methods of analysis. Chem. Phys. Lipids 2018, 216, 114–131.
- Barrera, N.P.; Zhou, M.; Robinson, C.V. The role of lipids in defining membrane protein interactions: Insights from mass spectrometry. Trends Cell Biol. 2013, 23, 1–8.
- Bollinger, C.R.; Teichgräber, V.; Gulbins, E. Ceramide-enriched membrane domains. Biochim. Biophys. Acta BBA Bioenerg. 2005, 1746, 284–294.
- Gulbins, E.; Kolesnick, R. Raft ceramide in molecular medicine. Oncogene 2003, 22, 7070–7077.
- Bedia, C.; Levade, T.; Codogno, P. Regulation of Autophagy by Sphingolipids. Anti-Cancer Agents Med. Chem. 2011, 11, 844–853.
- Young, M.; Kester, M.; Wang, H.-G. Sphingolipids: Regulators of crosstalk between apoptosis and autophagy. J. Lipid Res. 2013, 54, 5–19.
- Carpinteiro, A.; Dumitru, C.; Schenck, M.; Gulbins, E. Ceramide-induced cell death in malignant cells. Cancer Lett. 2008, 264, 1–10.
- Weigert, A.; Olesch, C.; Brüne, B. Sphingosine-1-Phosphate and Macrophage Biology—How the Sphinx Tames the Big Eater. Front. Immunol. 2019, 10, 1706.
- Alvarez, S.E.; Harikumar, K.B.; Hait, N.C.; Allegood, J.; Strub, G.M.; Kim, E.Y.; Maceyka, M.; Jiang, H.; Luo, C.; Kordula, T.; et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 2010, 465, 1084–1088.
- Smith, E.L.; Schuchman, E.H. The unexpected role of acid sphingomyelinase in cell death and the pathophysiology of common diseases. FASEB J. 2008, 22, 3419–3431.
- Schulze, H.; Sandhoff, K. Lysosomal Lipid Storage Diseases. Cold Spring Harb. Perspect. Biol. 2011, 3, a004804.
- Kornhuber, J.; Müller, C.; Becker, K.A.; Reichel, M.; Gulbins, E. The ceramide system as a novel antidepressant target. Trends Pharmacol. Sci. 2014, 35, 293–304.
- Halmer, R.; Walter, S.; Fassbender, K. Sphingolipids: Important Players in Multiple Sclerosis. Cell. Physiol. Biochem. 2014, 34, 111–118.
- Feng, S.; Harayama, T.; Montessuit, S.; David, F.P.; Winssinger, N.; Martinou, J.-C.; Riezman, H. Mitochondria-specific photoactivation to monitor local sphingosine metabolism and function. eLife 2018, 7, 7.
- Sakamoto, W.; Canals, D.; Salamone, S.; Allopenna, J.; Clarke, C.J.; Snider, J.; Obeid, L.M.; Hannun, Y.A. Probing compartment-specific sphingolipids with targeted bacterial sphingomyelinases and ceramidases. J. Lipid Res. 2019, 60, 1841–1850.
- Höglinger, D.; Nadler, A.; Haberkant, P.; Kirkpatrick, J.; Schifferer, M.; Stein, F.; Hauke, S.; Porter, F.D.; Schultz, C. Trifunctional lipid probes for comprehensive studies of single lipid species in living cells. Proc. Natl. Acad. Sci. USA 2017, 114, 1566–1571.
- Contreras, F.-X.; Ernst, A.; Haberkant, P.; Björkholm, P.; Lindahl, E.; Gönen, B.; Tischer, C.; Elofsson, A.; von Heijne, G.; Thiele, C.; et al. Molecular recognition of a single sphingolipid species by a protein’s transmembrane domain. Nature 2012, 481, 525–529.
- Contreras, F.-X.; Ernst, A.; Wieland, F.; Brügger, B. Specificity of Intramembrane Protein-Lipid Interactions. Cold Spring Harb. Perspect. Biol. 2011, 3, a004705.
- Tamura, T.; Fujisawa, A.; Tsuchiya, M.; Shen, Y.; Nagao, K.; Kawano, S.; Tamura, Y.; Endo, T.; Umeda, M.; Hamachi, I. Organelle membrane-specific chemical labeling and dynamic imaging in living cells. Nat. Chem. Biol. 2020, 16, 1361–1367.
- Kunz, T.C.; Kozjak-Pavlovic, V. Diverse Facets of Sphingolipid Involvement in Bacterial Infections. Front. Cell Dev. Biol. 2019, 7, 203.
- Rolando, M.; Buchrieser, C. A Comprehensive Review on the Manipulation of the Sphingolipid Pathway by Pathogenic Bacteria. Front. Cell Dev. Biol. 2019, 7, 168.
- Li, C.; Wang, A.; Wu, Y.; Gulbins, E.; Grassmé, H.; Zhao, Z. Acid Sphingomyelinase-Ceramide System in Bacterial Infections. Cell. Physiol. Biochem. 2019, 52, 280–301.
- Becker, K.A.; Gellhaus, A.; Winterhager, E.; Gulbins, E. Ceramide-Enriched Membrane Domains in Infectious Biology and Development. Alzheimer’s Dis. 2008, 49, 523–538.
- Fink, J.; Schumacher, F.; Schlegel, J.; Stenzel, P.; Wigger, D.; Sauer, M.; Kleuser, B.; Seibel, J. Azidosphinganine enables metabolic labeling and detection of sphingolipid de novo synthesis. Org. Biomol. Chem. 2021, 19, 2203–2212.
- Wigger, D.; Gulbins, E.; Kleuser, B.; Schumacher, F. Monitoring the Sphingolipid de novo Synthesis by Stable-Isotope Labeling and Liquid Chromatography-Mass Spectrometry. Front. Cell Dev. Biol. 2019, 7, 210.
- Doktorova, M.; Symons, J.L.; Levental, I. Structural and functional consequences of reversible lipid asymmetry in living membranes. Nat. Chem. Biol. 2020, 16, 1321–1330.
- Airola, M.V.; Hannun, Y.A. Sphingolipid Metabolism and Neutral Sphingomyelinases. Handb. Exp. Pharmacol. 2013, 2013, 57–76.
- Clarke, C.J.; Snook, C.F.; Tani, M.; Matmati, N.; Marchesini, A.N.; Hannun, Y.A. The Extended Family of Neutral Sphingomyelinases. Biochemistry 2006, 45, 11247–11256.
- Airola, M.; Shanbhogue, P.; Shamseddine, A.A.; Guja, K.E.; Senkal, C.E.; Maini, R.; Bartke, N.; Wu, B.X.; Obeid, L.M.; Garcia-Diaz, M.; et al. Structure of human nSMase2 reveals an interdomain allosteric activation mechanism for ceramide generation. Proc. Natl. Acad. Sci. USA 2017, 114, E5549–E5558.
- Avota, E.; De Lira, M.N.; Schneider-Schaulies, S. Sphingomyelin Breakdown in T Cells: Role of Membrane Compartmentalization in T Cell Signaling and Interference by a Pathogen. Front. Cell. Dev. Biol. 2019, 7, 152.
- Andrews, N.W. Solving the secretory acid sphingomyelinase puzzle: Insights from lysosome-mediated parasite invasion and plasma membrane repair. Cell. Microbiol. 2019, 21, e13065.
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An Overview of Sphingolipid Metabolism: From Synthesis to Breakdown. Sphingolipids Signal. Regul. Mol. 2010, 1–23.
- Spiegel, S.; Maczis, M.A.; Maceyka, M.; Milstien, S. New insights into functions of the sphingosine-1-phosphate transporter SPNS2. J. Lipid Res. 2019, 60, 484–489.
- Takabe, K.; Spiegel, S. Export of sphingosine-1-phosphate and cancer progression. J. Lipid Res. 2014, 55, 1839–1846.
- Dissanayake, T.K.; Yan, B.; Ng, A.C.-K.; Zhao, H.; Chan, G.; Yip, C.C.-Y.; Sze, K.-H.; To, K.K.-W. Differential role of sphingomyelin in influenza virus, rhinovirus and SARS-CoV-2 infection of Calu-3 cells. J. Gen. Virol. 2021, 102, 001593.
- Soudani, N.; Hage-Sleiman, R.; Karam, W.; Dbaibo, G.; Zaraket, H. Ceramide Suppresses Influenza A Virus Replication In Vitro. J. Virol. 2019, 93.
- Tatematsu, K.; Tanaka, Y.; Sugiyama, M.; Sudoh, M.; Mizokami, M. Host sphingolipid biosynthesis is a promising therapeutic target for the inhibition of hepatitis B virus replication. J. Med. Virol. 2011, 83, 587–593.
- Perera, R.; Riley, C.; Isaac, G.; Hopf-Jannasch, A.S.; Moore, R.J.; Weitz, K.W.; Pasa-Tolic, L.; Metz, T.; Adamec, J.; Kuhn, R.J. Dengue Virus Infection Perturbs Lipid Homeostasis in Infected Mosquito Cells. PLoS Pathog. 2012, 8, e1002584.
- Zhang, Z.; He, G.; Filipowicz, N.A.; Randall, G.; Belov, G.A.; Kopek, B.G.; Wang, X. Host Lipids in Positive-Strand RNA Virus Genome Replication. Front. Microbiol. 2019, 10, 286.
- Perera, M.N.; Ganesan, V.; Siskind, L.J.; Szulc, Z.M.; Bielawski, J.; Bielawska, A.; Bittman, R.; Colombini, M. Ceramide channels: Influence of molecular structure on channel formation in membranes. Biochim. Biophys. Acta BBA Biomembr. 2012, 1818, 1291–1301.
- Sengupta, P.; Lippincott-Schwartz, J. Revisiting Membrane Microdomains and Phase Separation: A Viral Perspective. Viruses 2020, 12, 745.
- Sengupta, P.; Seo, A.Y.; Pasolli, H.A.; Song, Y.E.; Johnson, M.C.; Lippincott-Schwartz, J. A lipid-based partitioning mechanism for selective incorporation of proteins into membranes of HIV particles. Nat. Cell Biol. 2019, 21, 452–461.
- Brügger, B.; Glass, B.; Haberkant, P.; Leibrecht, I.; Wieland, F.T.; Kräusslich, H.-G. The HIV lipidome: A raft with an unusual composition. Proc. Natl. Acad. Sci. USA 2006, 103, 2641–2646.
- Lorizate, M.; Sachsenheimer, T.; Glass, B.; Habermann, A.; Gerl, M.; Kräusslich, H.-G.; Brügger, B. Comparative lipidomics analysis of HIV-1 particles and their producer cell membrane in different cell lines. Cell. Microbiol. 2013, 15, 292–304.
- Favard, C.; Chojnacki, J.; Merida, P.; Yandrapalli, N.; Mak, J.; Eggeling, C.; Muriaux, D. HIV-1 Gag specifically restricts PI(4,5)P2 and cholesterol mobility in living cells creating a nanodomain platform for virus assembly. Sci. Adv. 2019, 5, eaaw8651.
- Briggs, J.A.G.; Riches, J.; Glass, B.; Bartonova, V.; Zanetti, G.; Krausslich, H.-G. Structure and assembly of immature HIV. Proc. Natl. Acad. Sci. USA 2009, 106, 11090–11095.
- Carlson, L.-A.; Briggs, J.; Glass, B.; Riches, J.; Simon, M.N.; Johnson, M.C.; Müller, B.; Grünewald, K.; Kräusslich, H.-G. Three-Dimensional Analysis of Budding Sites and Released Virus Suggests a Revised Model for HIV-1 Morphogenesis. Cell Host Microbe 2008, 4, 592–599.
- Pastenkos, G.; Miller, J.L.; Pritchard, S.M.; Nicola, A.V. Role of Sphingomyelin in Alphaherpesvirus Entry. J. Virol. 2019, 93.
- Audi, A.; Soudani, N.; Dbaibo, G.; Zaraket, H. Depletion of Host and Viral Sphingomyelin Impairs Influenza Virus Infection. Front. Microbiol. 2020, 11, 612.
- Perez-Zsolt, D.; Erkizia, I.; Pino, M.; García-Gallo, M.; Martin, M.T.; Benet, S.; Chojnacki, J.; Fernández-Figueras, M.T.; Guerrero, D.; Urrea, V.; et al. Anti-Siglec-1 antibodies block Ebola viral uptake and decrease cytoplasmic viral entry. Nat. Microbiol. 2019, 4, 1558–1570.
- Puryear, W.B.; Akiyama, H.; Geer, S.D.; Ramirez, N.-G.; Yu, X.; Reinhard, B.M.; Gummuluru, S. Interferon-Inducible Mechanism of Dendritic Cell-Mediated HIV-1 Dissemination Is Dependent on Siglec-1/CD169. PLoS Pathog. 2013, 9, e1003291.
- Resop, R.S.; Fromentin, R.; Newman, D.; Rigsby, H.; Dubrovsky, L.; Bukrinsky, M.; Chomont, N.; Bosque, A. Fingolimod inhibits multiple stages of the HIV-1 life cycle. PLoS Pathog. 2020, 16, e1008679.
- Studstill, C.J.; Pritzl, C.J.; Seo, Y.-J.; Kim, D.Y.; Xia, C.; Wolf, J.J.; Nistala, R.; Vijayan, M.; Cho, Y.-B.; Kang, K.W.; et al. Sphingosine kinase 2 restricts T cell immunopathology but permits viral persistence. J. Clin. Investig. 2020, 130, 6523–6538.
- Naz, F.; Arish, M. Battling COVID-19 Pandemic: Sphingosine-1-Phosphate Analogs as an Adjunctive Therapy? Front. Immunol. 2020, 11, 1102.
- Walsh, K.B.; Teijaro, J.R.; Brock, L.G.; Fremgen, D.M.; Collins, P.L.; Rosen, H.; Oldstone, M.B.A. Animal Model of Respiratory Syncytial Virus: CD8+ T Cells Cause a Cytokine Storm That Is Chemically Tractable by Sphingosine-1-Phosphate 1 Receptor Agonist Therapy. J. Virol. 2014, 88, 6281–6293.
- Walsh, K.B.; Teijaro, J.R.; Wilker, P.R.; Jatzek, A.; Fremgen, D.M.; Das, S.C.; Watanabe, T.; Hatta, M.; Shinya, K.; Suresh, M.; et al. Suppression of cytokine storm with a sphingosine analog provides protection against pathogenic influenza virus. Proc. Natl. Acad. Sci. USA 2011, 108, 12018–12023.
- McGowan, E.M.; Haddadi, N.; Nassif, N.T.; Lin, Y. Targeting the SphK-S1P-SIPR Pathway as a Potential Therapeutic Approach for COVID-19. Int. J. Mol. Sci. 2020, 21, 7189.
- Teijaro, J.R.; Studer, S.; Leaf, N.; Kiosses, W.B.; Nguyen, N.; Matsuki, K.; Negishi, H.; Taniguchi, T.; Oldstone, M.B.A.; Rosen, H. S1PR1-mediated IFNAR1 degradation modulates plasmacytoid dendritic cell interferon-α autoamplification. Proc. Natl. Acad. Sci. USA 2016, 113, 1351–1356.
- Vijayan, M.; Xia, C.; Song, Y.E.; Ngo, H.; Studstill, C.J.; Drews, K.; Fox, T.E.; Johnson, M.C.; Hiscott, J.; Kester, M.; et al. Sphingosine 1-Phosphate Lyase Enhances the Activation of IKKepsilon To Promote Type I IFN-Mediated Innate Immune Responses to Influenza A Virus Infection. J. Immunol. 2017, 199, 677–687.
- Verderio, C.; Gabrielli, M.; Giussani, P. Role of sphingolipids in the biogenesis and biological activity of extracellular vesicles. J. Lipid Res. 2018, 59, 1325–1340.
- Dinkins, M.B.; Wang, G.; Bieberich, E. Sphingolipid-Enriched Extracellular Vesicles and Alzheimer’s Disease: A Decade of Research. J. Alzheimer’s Dis. 2017, 60, 757–768.
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brugger, B.; Simons, M. Ceramide Triggers Budding of Exosome Vesicles into Multivesicular Endosomes. Science 2008, 319, 1244–1247.
- Catalano, M.; O’Driscoll, L. Inhibiting extracellular vesicles formation and release: A review of EV inhibitors. J. Extracell. Vesicles 2020, 9, 1703244.
- Raab-Traub, N.; Dittmer, D.P. Viral effects on the content and function of extracellular vesicles. Nat. Rev. Microbiol. 2017, 15, 559–572.
- Li, J.; Liu, K.; Liu, Y.; Xu, Y.; Zhang, F.; Yang, H.; Liu, J.; Pan, T.; Chen, J.; Wu, M.; et al. Exosomes mediate the cell-to-cell transmission of IFN-α-induced antiviral activity. Nat. Immunol. 2013, 14, 793–803.