Alternative splicing of mRNA is an essential mechanism to regulate and increase the diversity of the transcriptome and proteome. Alternative splicing frequently occurs in a tissue- or time-specific manner, contributing to differential gene expression between cell types during development. Neural tissues present extremely complex splicing programs and display the highest number of alternative splicing events. As an extension of the central nervous system, the retina constitutes an excellent system to illustrate the high diversity of neural transcripts. The retina expresses retinal specific splicing factors and produces a large number of alternative transcripts, including exclusive tissue-specific exons, which require an exquisite regulation. In fact, a current challenge in the genetic diagnosis of inherited retinal diseases stems from the lack of information regarding alternative splicing of retinal genes, as a considerable percentage of mutations alter splicing or the relative production of alternative transcripts. Modulation of alternative splicing in the retina is also instrumental in the design of novel therapeutic approaches for retinal dystrophies, since it enables precision medicine for specific mutations.
- retina
- alternative splicing
- inherited retinal dystrophies
- splicing factors
- microexons
Introduction
The human genome is estimated to contain ∼30,000 genes, which represent about 1% of the total genome[1]. Although the number of genes is not much higher than in invertebrates, a higher molecular complexity can be attained by the production of multiple mRNA isoforms from a single gene, generated by alternative splicing (AS) and leading to a huge diversification of the proteome[2]. Alternative splicing events (ASEs) of pre-mRNA transcripts strongly contribute to the cellular regulatory landscape, protein diversity, and therefore, to organismal complexity, together with other related mechanisms such as the use of alternative promoters, transcription start sites and polyadenylation sites. AS consists in the selective removal or retention of introns and exons giving rise to different rearranged patterns of mature mRNA products. The proper recognition of pre-mRNA regions during splicing is mediated by cis-acting regulatory elements, whose location on the pre-mRNA spatially orchestrates the splicing event. In addition, cis-regulatory sequences can interact with trans- acting regulatory elements to promote or repress splicing. The most common occurrence of ASEs is observed in the central nervous system (CNS) of vertebrates[3], where it plays pivotal roles in processes such as neurogenesis, cell migration, synaptogenesis, synaptic function or neuronal network function and plasticity[4]. The retina, the light sensing tissue of the eye, is a highly specialized tissue belonging to the CNS. High levels of ASEs occur in the retina where precise gene regulation is required for neuronal development and function. The importance of AS in retina is highlighted by numerous examples where splicing alterations underly retinal disorders and disease, such as cone-rod dystrophy, Usher syndrome (USH) and retinitis pigmentosa (RP). Notably, splicing mutations account for 9% of all disease-causing mutations reported in the Human Gene Mutation Database (HGMD)[5]. These mutations are hard to identify and are often miscategorized as missense, nonsense, or silent, although they should be considered as splicing mutations.
Alternative Splicing of Retinal Genes
In the retina, AS represents a crucial regulatory step of gene expression during development and homeostasis. Many different types of AS have been identified in the retina (Figure 1), including cassette exons[6], exon skipping[7] and intron retention events[8], mutually exclusive exons[9], alternative splice sites[10], alternative promoters[11] and alternative polyadenylation (polyA) sites[12]. Interestingly, a specialized type of splicing leading to incorporation of alternative microexons (exons that are ≤30 nt) has been shown to impact neuronal differentiation and function[13].
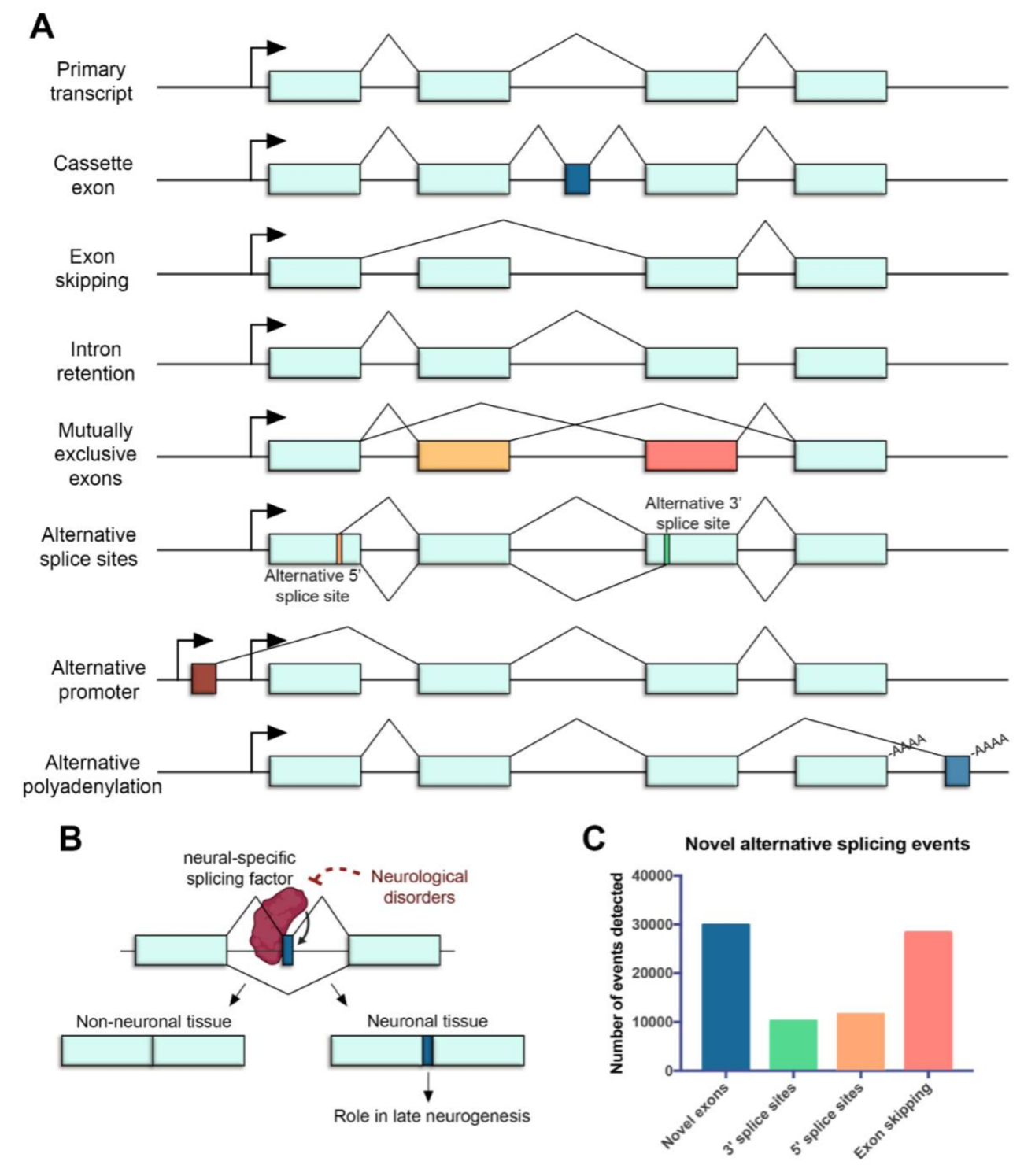
Figure 1. Alternative splicing in the neural retina. (A) Common mechanisms of alternative splicing in the retina. Boxes represent exons, lines represent introns, promoters are represented with arrows and polyadenylation sites are indicated with -AAAA. Exon regions included in the alternative transcript are colored. (B) Microexons have a role in late neurogenesis and are relevant in neurological disorders. The reduced expression of neural-specific splicing factors that regulate the inclusion of microexons is linked to the altered splicing of microexons in patients with neurological disorders. (C) Novel alternative splicing events in the human retina detected by RNA sequencing (data from [14]).
The splicing process is carried out by a dynamic ribonucleoprotein (RNP) machinery, the spliceosome, which is characterized by the orchestrated assembly and disassembly of several small nuclear RNPs (snRNPs) and associated protein co-factors (Figure 2). Fundamentally, the spliceosomal core is comprised of five different snRNPs named U1, U2, U4, U5 and U6 after the small nuclear RNAs (snRNAs) that compose them. mutations in splicing factors ubiquitously expressed and important for the general process of pre-mRNA splicing can cause a retina- restricted phenotype. Interestingly, most of these factors are essential for the interaction between U4/U6 and U5 snRNPs and the stabilization of the U4/U6·U5 tri-snRNP, such as PRPF3, PRPF4, PRPF6, PRPF8 and PRPF31. Therefore, proper splicing of retinal transcripts is an important process for accurate retinal function and homeostasis, making the retina particularly sensitive to splicing alterations.
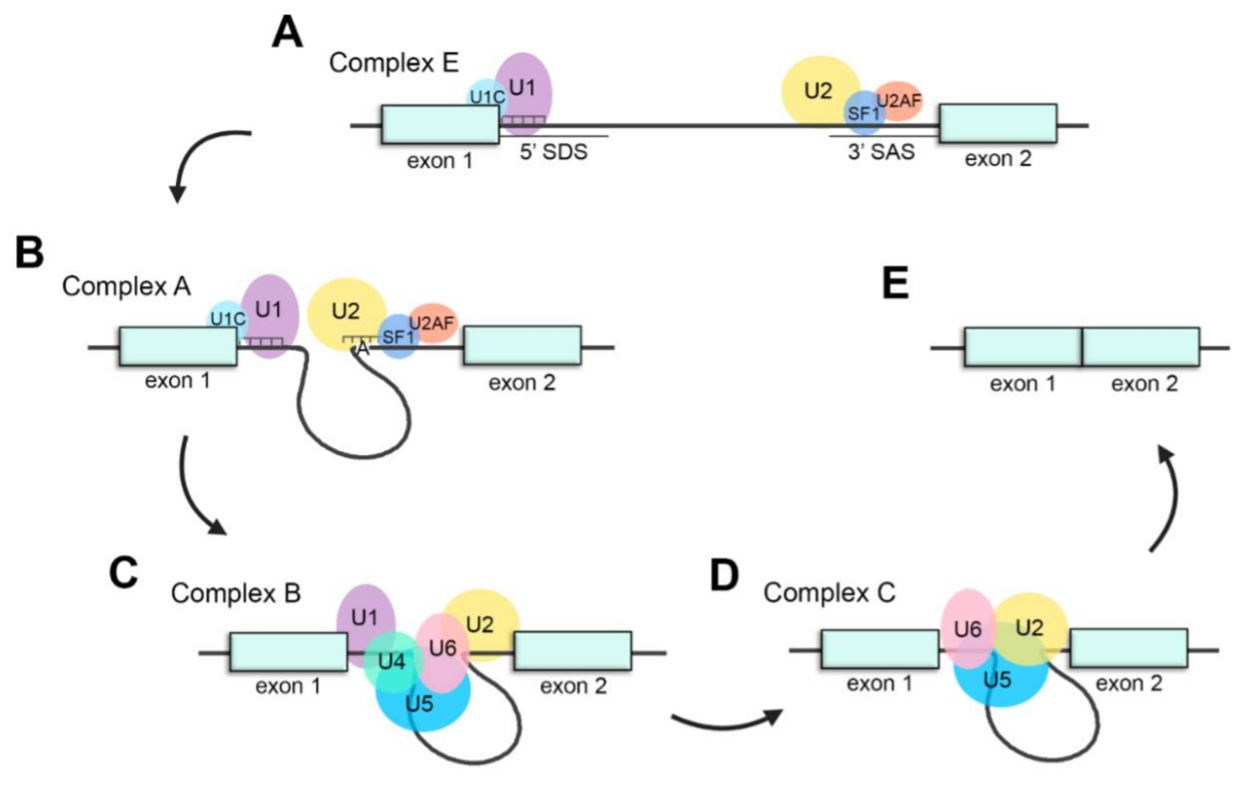
Figure 2. Schematic representation of the splicing process. (A) Assembly of the spliceosome: U1 snRNP recognizes the splice donor site (SDS) and U2 snRNP recognizes the splice acceptor site (SAS) to generate complex E. (B) U2 recognizes the adenosine at the branch-site and forms complex A. (C) The U4/U6·U5 tri-snRNP joins the spliceosome to form complex B. (D) U4 and U1 are released, U6 replaces U1 recognizing the SDS and interacts with U2, generating complex C and catalyzing the splicing reaction. (E) Exons are ligated, and intronic pre-mRNA and spliceosomal snRNPs are liberated.
The Role of Alternative Splicing in Retinal Disease
Around 9% of all disease-causing mutations are estimated to alter pre-mRNA splicing[15]. These mutations can disrupt or alter cis-regulatory sequences, or the binding of trans-acting splicing factors. Mutations affecting cis-acting splice sites or regulatory sequences can lead to inappropriate exon skipping, intron inclusion, exon inclusion or activation of cryptic splice sites, usually leading to frameshifts and premature termination. Mutations causing IRDs can also affect unannotated isoforms, which may cause misinterpretation of the genetic diagnosis. Around 90% of the IRD genes produce more than one transcript (Figure 3A), and for most of them (54%), between two to 10 alternatively spliced transcripts have been reported. Indeed, the multiplicity of transcripts also impacts on the multiplicity of encoded proteins, and at least 85% of the IRD genes display several protein isoforms (Figure 3B). Most of the genes produce between two and five distinct coding transcripts, but 9% of them can generate more than 10 singular coding transcripts that translate into proteins that most probably carry out differential functions. Therefore, understanding AS and isoform sequence information is fundamental to comprehend both normal gene function and the phenotypic consequences of gene mutations.
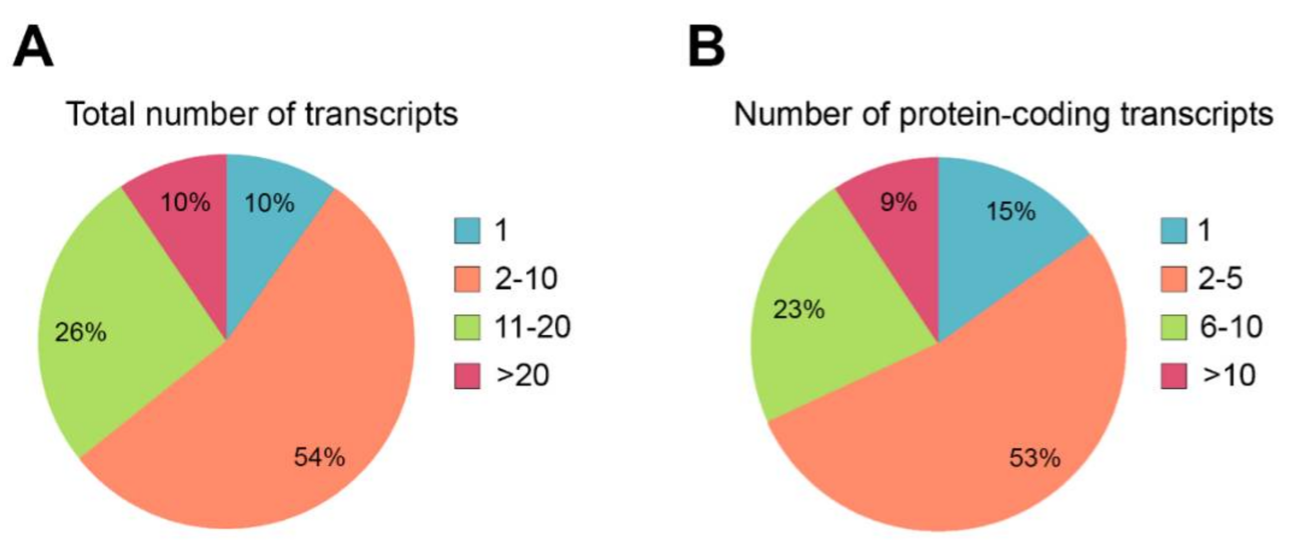
Figure 3. Quantification of alternative splicing events described in genes causing IRDs. (A) Percentage of IRD genes presenting alternative splicing events. Only 10% of the IRD genes produces a unique transcript, and most genes (54%) generate between 2 and 10 transcripts. (B) Percentage of IRD genes showing diverse protein-coding transcripts. Only 15% of the IRD genes produce one protein isoform. Most of the genes (53%) produce between 2 and 5 diverse protein isoforms.
Splice-site mutations have been identified in patients with RP, USH or Stargardt disease, among other IRDs. Mutations can either disrupt a consensus splice site sequence causing exon skipping, shift the splicing acceptor or donor splices sites, or promote the usage of cryptic deep intronic sequences that result in different exon size, intron retention, or novel exon inclusion. For instance, Non-Canonical Splice Site Variants (NCSS) may alter the splicing motif recognition by the splicing factors, which can lead to partial or entire exon skipping, producing partial in-frame deletions or open reading frame disruptions that cause frameshifts and lead to prematurely truncated proteins. Deep intronic mutations are located more than 100 bp away from exon-intron junctions and usually lead to pseudo-exon inclusion due to activation of novel splice sites. The introduction of a pseudo-exon (PE) commonly alters the reading frame and result in a premature stop codon that targets the mutant mRNA for degradation by non-sense mediated decay (NMD)[16]. Interestingly, mutations located within an exon can also affect splicing if they generate a strong donor or acceptor site. Therefore, point mutations located in exons should be routinely evaluated in silico and subsequently tested for their potential disruptive effect in mRNA splicing in order to avoid misinterpretation of the variants as well as to understand genotype-phenotype correlations, disease mechanisms and ultimately predict the disease course. Some mutations in widely expressed genes can result in primary ocular diseases if they alter retina-specific exons or affect the prevalence of retina-specific transcripts[17].
In conclusion, identifying retina-specific transcripts is essential to increase the genetic diagnosis yield in IRDs as well as to design precision medicine therapeutic approaches for specific mutations, based on RNA splicing modulation.
References
- Patrushev, L.I.; Kovalenko, T.F. Functions of noncoding sequences in mammalian genomes. Biochemistry 2014, 79, 1442–1469.
- Liu, Y.; Gonzàlez-Porta, M.; Santos, S.; Brazma, A.; Marioni, J.C.; Aebersold, R.; Venkitaraman, A.R.; Wickramasinghe, V.O. Impact of Alternative Splicing on the Human Proteome. Cell Rep. 2017, 20, 1229–1241.
- Furlanis, E.; Scheiffele, P. Regulation of Neuronal Differentiation, Function, and Plasticity by Alternative Splicing. Annu. Rev. Cell Dev. Biol. 2018, 34, 451–469.
- Hermey, G.; Blüthgen, N.; Kuhl, D. Neuronal activity-regulated alternative mRNA splicing. Int. J. Biochem. Cell Biol. 2017, 91, 184–193.
- Anna, A.; Monika, G. Splicing mutations in human genetic disorders: Examples, detection, and confirmation. J. Appl. Genet. 2018, 59, 253–268.
- Wan, J.; Masuda, T.; Hackler, L.; Torres, K.M.; Merbs, S.L.; Zack, D.J.; Qian, J. Dynamic usage of alternative splicing exons during mouse retina development. Nucleic Acids Res. 2011, 39, 7920–7930.
- Fong, H.K.W.; Lin, M.Y.; Pandey, S. Exon-skipping variant of RGR opsin in human retina and pigment epithelium. Exp. Eye Res. 2006, 83, 133–140.
- Aísa-Marín, I.; López-Iniesta, M.J.; Milla, S.; Lillo, J.; Navarro, G.; de la Villa, P.; Marfany, G. Nr2e3 functional domain ablation by CRISPR-Cas9D10Aidentifies a new isoform and generates retinitis pigmentosa and enhanced S-cone syndrome models. Neurobiol. Dis. 2020, 146.
- Mellough, C.B.; Bauer, R.; Collin, J.; Dorgau, B.; Zerti, D.; Dolan, D.W.P.; Jones, C.M.; Izuogu, O.G.; Yu, M.; Hallam, D.; et al. An integrated transcriptional analysis of the developing human retina. Development 2019, 146.
- Stojic, J.; Stöhr, H.; Weber, B.H.F. Three novel ABCC5 splice variants in human retina and their role as regulators of ABCC5 gene expression. BMC Mol. Biol. 2007, 8.
- Campla, C.K.; Mast, H.; Dong, L.; Lei, J.; Halford, S.; Sekaran, S.; Swaroop, A. Targeted deletion of an NRL- and CRX-regulated alternative promoter specifically silences FERM and PDZ domain containing 1 (Frmpd1) in rod photoreceptors. Hum. Mol. Genet. 2019, 28, 804–817.
- Hu, W.; Li, S.; Park, J.Y.; Boppana, S.; Ni, T.; Li, M.; Zhu, J.; Xie, Z.; Xiang, M. Dynamic landscape of alternative polyadenylation during retinal development HHS Public Access. Cell Mol. Life Sci. 2017, 74, 1721–1739.
- Yang, L.; Chen, L.L. Microexons go big. Cell 2014, 159, 1488–1489.
- Farkas, M.H.; Grant, G.R.; White, J.A.; Sousa, M.E.; Consugar, M.B.; Pierce, E.A. Transcriptome analyses of the human retina identify unprecedented transcript diversity and 3.5 Mb of novel transcribed sequence via significant alternative splicing and novel genes. BMC Genom. 2013, 14.
- Bergsma, A.J.; van der Wal, E.; Broeders, M.; van der Ploeg, A.T.; Pim Pijnappel, W.W.M. Alternative Splicing in Genetic Diseases: Improved Diagnosis and Novel Treatment Options. Int. Rev. Cell Mol. Biol. 2018, 335, 85–141.
- Vaz-Drago,R.;Custódio,N.;Carmo-Fonseca,M.Deepintronicmutationsandhumandisease.Hum.Genet.2017,136,1093–1111.
- Liu, M.M.; Zack, D.J. Alternative splicing and retinal degeneration. Clin. Genet. 2013, 84, 142–149.