Nucleotide-binding oligomerization domain NOD-like receptors (NLRs) are conserved cytosolic pattern recognition receptors (PRRs) that track the intracellular milieu for the existence of infection, disease-causing microbes, as well as metabolic distresses. The NLRP3 inflammasome agglomerates are consequent to sensing a wide spectrum of pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs). Certain members of the NLR family have been documented to lump into multimolecular conglomerates called inflammasomes, which are inherently linked to stimulation of the cysteine protease caspase-1.
- NOD-like receptors
- NLRP3
- inflammasome
- inflammation
- disease inhibitor
1. Introduction
Nucleotide-binding oligomerization domain NOD-like receptors (NLRs) are preserved pattern recognition receptors located in cell cytosol that track the intracellular milieu for the presence of infection, disease-causing microbes, as well as metabolic distresses. In humans, the NLR family is made up of 23 cytosolic proteins, and some 34 nlr murine genes have been determined. The usual segment structure of the NLR family members comprises of an amino-terminal effector part made up of a protein–protein interaction region, like the caspase-recruitment domain (CARD), pyrin domain (PYD), or Baculovirus inhibitor of apoptosis protein repeat (BIR) segment, a centrally positioned NOD domain and leucine-rich repeats associated with danger sensing at the carboxyl-terminal. Alterations at the N-terminal domain are employed for the subsequent assortment of NLR protein members. The biggest assortment incorporates the N-terminal PYD and has been christened the NLRPs. Certain members of the NLR family have been known to agglomerate into multimolecular structures called inflammasomes, which are inherently linked to the stimulation of the cysteine protease caspase-1. Following activation, caspase-1 severs the proinflammatory cytokines interleukin (IL)-1β and IL-18 into their biologically active entities, leading to the commencement of caspase-1-associated pyroptosis. IL-1β is an archetypic inflammatory cytokine implicated in multiple types of inflammatory maladies. Approaches to impede IL-1β ’s actions are possible, and their therapeutic effects have been demonstrated; nevertheless, such strategies are associated with certain constraints. For instance, treatments that focus on systemically negating IL-1β (i.e., anakinra, rilonacept, and canakinumab) have been reported to subsequently result in an escalated peril of infections and are hence deemed improper for oral use. NLRP3 is the most common inflammasome sensor probed for its association in a multitude of conditions, such as sterile inflammation, infections, as well as uncommon genetic autoimmune syndromes ( Table 1 ).
Table 1. Some inflammatory diseases with their targets (involving inflammasome signaling components) for possible therapeutic intervention.
| Disease | Targets in Inflammasome Signaling Cascade | Therapeutic Molecule |
|---|---|---|
| Acute Myocardial Infarction | NLRP3 | Colchicine |
| Type 2 Diabetes Mellitus | NLRP3(indirect action) | Metformin, Glyburide |
| IL-1β | Rilonacept | |
| Rheumatoid Arthritis | IL-1 Receptor | Anakinra |
| Caspase-1 | Pralnacasan(VX-740) | |
| P2X7 | AZD9056, CE-224535, GSK 1482169 | |
| Muckle–Wells Syndrome | Caspase-1 | Emricasan(VX-765) |
| IL-1β | Canakinumab | |
| Gout | IL-1β | Rilonacept |
| Xanthine Oxidase(XOD) | Allopurinol | |
| Systemic Lupus Erythematosus | NFκB (IKKβ kinase activity)/NLRP3 ATPase | Bay 11-7082 |
| Cryopyrin-Associated Periodic Syndromes(CAPS) | IL-1β | Rilonacept |
| Inflammatory Bowel Disease(IBD) | IL-18 | GSK1070806 |
| Familial Cold Autoinflammatory Syndrome(FCAS) | IL-1β | Canakinumab |
| Cancer | Caspase-1/NF-κB (IKKβ kinase activity)/NLRP3 ATPase | Parthenolide |
| B-cell Non-Hodgkin’s Lymphoma | IL-18 | GSK1070806 |
Given the therapeutic promise of an NLRP3 inhibitor, the concerted escalated venture of the scientific fraternity in the yesteryears towards the development of small molecules focusing on NLRP3 is quite predictable. However, incomplete comprehension of the steps leading to the NLRP3 inflammasome agglomeration and also insufficient understanding of the sensor crystal structure compound the odds of developing such inhibitor agents. Despite the fact that certain NLRP3 inflammasome antagonists have been developed and studied in preclinical protocols as well as cell-based assays, an NLRP3-specific inhibitor with therapeutic intent for humans has yet to be licensed. This review is intended to showcase the current developments with regard to promising NLRP3 inhibitors for clinical applications ( Table 2 ).
Table 2. Targets of some known NLRP3 Inhibitors.
| Inhibitor | Target(s) | Documented Mechanism(s) | References | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sulfonylureas | ||||||||||||
| Glyburide | NLRP3(indirect action) | Abrogation of ASC agglomeration acting downstream of P2X7; Suppression of K | ATP | channels | [1] | |||||||
| MCC950 | NLRP3 | NLRP3 inflammasome activation involves a role of its ATPase domain. MCC950 is known to directly target and restrain this ATP-hydrolysis motif in both canonical as well as non-canonical NLRP3 inflammasomes | [2][3] | [2,3] | ||||||||
| Glitazones | ||||||||||||
| CY-09 | NLRP3 | Effective and direct suppressor of NLRP3 inflammasome with remarkable capability to impede NLRP3 inflammasome activation in vivo in murine models and ex vivo in human cells; blocks NLRP3 ATPase actions | [4] | |||||||||
| Substituted 2-pyrazolin-5-ones | ||||||||||||
| Edaravone | NLRP3 | Scavenge reactive oxygen species(ROS) thereby impeding NLRP3-evoked IL-1β processing and release; also known to suppress IL-1β, caspase 1 and NF-kB-reliant NLRP3 inflammation signaling | [5][6] | [5,6] | ||||||||
| Arsenic compounds | ||||||||||||
| Arsenic trioxide(As | 2 | O | 3 | ) | NLRP3 | As | 2 | O | 3 | suppresses NLRP3 inflammasome stimulation and consequent IL-1β and IL-18 release | [7][8] | [7,8] |
| Alkaloid | ||||||||||||
| Colchicine | NLRP3 | Efficaciously attenuates the expression levels of IL-1β, IL-6 and IL-18 by abrogating NLRP3 inflammasome activation cascade | [9][10] | [9,10] | ||||||||
| Biguanide | ||||||||||||
| Metformin | NLRP3 | Adenosine monophosphate-activated protein kinase(AMPK) is known to modulate NLRP3 inflammasome stimulation; decreases the expression of NLRP3 as well as kindling of the NLRP3 inflammasome signaling pathway | [11][12] | [11,12] | ||||||||
| GLP-1 analogs | ||||||||||||
| Liraglutide | NLRP3(hepatic) | Repression of the hepatic NLRP3 inflammasome | [13] | |||||||||
| Statins | ||||||||||||
| Atorvastatin | NLRP3 | Conspicuously decrements levels of NLRP3, caspase-1, and IL-1β; also, the NF-κB suppressor attenuate levels of inflammatory cytokines in inflammatory cells. The stimulation of the NF-κB signaling cascade is engaged in NLRP3 inflammasome activity modulation | [14][15] | [14,15] | ||||||||
| SGLT-2 Inhibitors(Dapagliflozin, Empagliflozin)-P2Y12 Antagonist(Ticagrelor) | ||||||||||||
| Dapagliflozin | NLRP3 | Extenuates inflammation-evoked renal damage and glomerulosclerosis in diabetic kidneys by ameliorating NLRP3 inflammasome stimulation; AMPK activation | [16] | |||||||||
| Empagliflozin | NLRP3 | Impedes kindling of NLRP3 inflammasome and decrements downstream inflammatory signaling in the diabetic kidneys | [17] | |||||||||
| Ticagrelor | NLRP3 | Repress NLRP3 inflammasome stimulation; AMPK activation | [18] | |||||||||
| Xanthine oxidase(XOD) enzyme inhibitor | ||||||||||||
| Allopurinol | NLRP3, XOD | Represses xanthine oxidase(XOD) action and subsequently attenuates generation of uric acid (UA) and reactive oxygen species (ROS), which are known to kindle the NLRP3 pathway | [19][20] | [19,20] | ||||||||
| Vinylsulfones | ||||||||||||
| BAY11-7082 | NLRP3, IKK, E2/3 enzymes, PTPs | Leads to cysteine alkylation of NLRP3 inflammasome ATPase domains; represses NLRP3 ATPase actions | [4][21] | [4,21] | ||||||||
| Beta-Nitrostyrenes | ||||||||||||
| MNS | NLRP3 | Leads to cysteine alteration of NLRP3 inflammasome ATPase domains; represses NLRP3 inflammasome actions | [22] | |||||||||
| Acrylate Derivatives | ||||||||||||
| INF39 | NLRP3 | Abrogates NLRP3 inflammasome ATPase actions; represses priming | [23] | |||||||||
| Acylhydrazone | ||||||||||||
| EMD638683 | NLRP3 | Suppression of NLRP3 and IL-1β expression | [24] | |||||||||
| Benzimidazoles | ||||||||||||
| FC11A-2 | NLRP3(indirect effect) | Hampers pro-caspase-1 autocleavage; impedes IL-1beta/18 secretion | [25][26] | [25,26] | ||||||||
| Sulfonylnitriles | ||||||||||||
| Dapansutrile(OLT1177) | NLRP3 | Abrogates NLRP3 inflammasome ATPase actions; suppresses NLRP3 inflammasome stimulation | [27][28] | [27,28] | ||||||||
| Benzoxathiole Derivatives | ||||||||||||
| BOT-4-one | NLRP3 | Akin to various covalent modulators that repress NLRP3, this agent blunts its ATPase activity; inhibits priming | [29][30][31] | [29,30,31] | ||||||||
| Tryptophan Derivative | ||||||||||||
| Tranilast | NLRP3 | Interacts with NACHT segment of NLRP3 to abrogate NLRP3-NLRP3 and NLRP3-ASC association | [4][32] | [4,32] | ||||||||
| Natural Products | ||||||||||||
| BHB | NLRP3(indirectly) | Abrogation of outward movement of K+ with consequent decrement in ASC agglomeration and IL-1beta/18 release | [33] | |||||||||
| Parthenolide | NLRP1 & 3, Caspase- 1, NF-kB, IKKB kinase activity | Alkyl modification of cysteine moieties present in ATPase segments of NLRP3 and caspase-1; abrogates NLRP3 ATPase actions | [34] | |||||||||
| Oridonin | NLRP3 | Selectively represses NLRP3 inflammasome stimulation; associates with cysteine 279 residue of NLRP3 and abrogates NLRP3-NEK7 association | [35] | |||||||||
| Caspase Inhibitors | ||||||||||||
| Pralnacasan(VX-740) | Caspase-1 | Covalent alteration of catalytic cysteine moiety in caspase-1 active site with consequent abrogation of caspase-1 effects and splitting of pro-IL-1Beta/18 | [36][37][38] | [36,37,38] | ||||||||
| Emricasan(VX-765) | Caspase-1 | Covalent alteration of catalytic cysteine moiety in caspase-1 active site with consequent abrogation of caspase-1 effects and splitting of pro-IL-1Beta/18 | [36][37][38] | [36,37,38] | ||||||||
2. NLRP3 Inflammasome Agglomeration
The NLRP3 inflammasome agglomerates are consequent to responding to a wide spectrum of pathogen-related molecular arrangements and damage-related molecular motifs. There is usually little cellular concentration of NLRP3, and in order to attain the critical threshold needed to spark caspase-1 activation, the canonical NLRP3 inflammasome stimulation hinges upon two crucial phases.
The first step, termed priming, leads to activation of nuclear factor kappa light chain enhancer of activated B cells (NF-kB) and various transcription factors following the involvement of pattern recognition receptors (PRRs). This engenders the expression of NLRP3 as well as pro-IL-1β. The second step consists of signal (or trigger) detection that in turn governs the triggering mechanism of NLRP3 and the subsequent generation of the inflammasome. The NLRP3 NACHT region, which possesses ATPase property, is essential for the agglomeration of the NLRP3 inflammasome. Following stimulation and self-oligomerization of NLRP3, the PYD–PYD cooperation among NLRP3 and the inflammasome adaptor protein apoptosis-associated speck-like protein having a CARD (ASC) leads to the generation of speck-like entities, which serves as a scaffold for the engagement by contiguity of procaspase-1 via CARD–CARD interaction. Following autocatalytic enzyme cleavage and the subsequent generation of active caspase-1 (p10 and p20), the transformation of pro-IL-1β and pro-IL-18 into their biologically active forms occurs and gasdermin D–moderated pyroptotic cell lethality ensues. Since a wide array of signals finally culminates in NLRP3 inflammasome generation, it is believed that NLRP3 can detect downstream developments arising from the archetype trigger that produces disruption of cellular homeostasis, with subsequent inflammasome agglomeration ( Figure 1 ). This disruption consists of alterations of ion flux (K + , Cl − , and Ca 2+ ), reactive oxygen species (ROS) generation, and lysosomal injury.
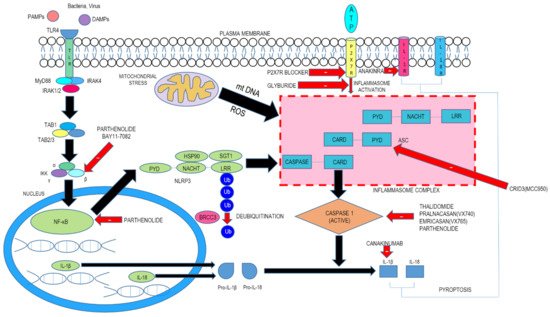
3. Inhibitors of NLRP3 Inflammasome-Driven In Vivo Disease Models
NLRP3 inflammasome brings about the sterile inflammatory reaction provoked by tissue injury and influences the causative pathology of myocardial ischemia–reperfusion injury [39][40][41][64,82,83]. Abrogating inflammasome stimulation by genetic method conspicuously attenuates infarct progress and myocardial fibrosis and malfunction [39][40][64,82].
In those afflicted with type 2 diabetes, enhanced NLRP3 inflammasome stimulation and processing of IL-1β were notably repressed by treatment with the antidiabetic drug metformin via AMPK stimulation [42][99].
The anticancer properties of BOT-4-one are mediated through the blockade of janus kinase 3 (JAK3)/signal transducer and activator of transcription 3 (STAT3) signaling [43][130] and immunomodulatory action via suppression of IKKb [44][131]. Upon testing for NF-kB-unrelated effects, BOT4-one was found to act akin to other alkylating agents like Bay 11-7082 and MNS, decrementing IL-1β release following canonical and noncanonical NLRP3 inflammasome stimulation, but with enhanced potency [29].
Scant information is available regarding the interaction of kinases with NLRP3; some other inflammasomes have been even less researched. Only if we can comprehend and dissect the kinase signaling networks encompassing these processes, their entire promise can be utilized, and pertinent kinase inhibitors may be ushered or repurposed into therapeutics as anti-inflammatory agents.
4. Conclusions
The intensive role of the inflammasome in masterminding innate immune responses (arising out of microbial infections and non-infectious diseases) has been proven beyond doubt by the occurrence of several heritable and acquired maladies which stem out of dysregulated NLRP3 inflammasome activation and the effectiveness of antagonists of IL-1β or its receptor for intervention in many of these disease conditions. NLRP3-induced pyroptosis and IL-1β/18 secretion are associated with numerous maladies. The degree to which NLRP3 inflammasome stimulation confers towards pyroptosis is still unclear, but NLRP3 activation does lead to pyroptosis, which consequently can inflict severe damage to crucial body organs [45][286]. Currently, NLRP3-linked afflictions may be ameliorated by agents which could abrogate IL-1β, such as counteracting IL-1β antibody canakinumab, recombinant IL-1 receptor antagonist anakinra, and the soluble camouflage IL-1 receptor, rilonacept. These biological molecules have been employed to manage cryopyrin-associated periodic syndromes (CAPS) as well as various maladies related to IL-1β [46][287]. Apart from NLRP-3 generated IL-1β, other cytokines like IL-18 may also assist with the development of the NLRP3-related afflictions [47][48][288,289]. Other inflammasomes or inflammasome-independent pathways can also lead to IL-1β production; hence, IL-1β inhibitors can only lead to non-intentional immunosuppressive actions. Thus, pharmacological inhibitors focusing on NLRP3 inflammasome inhibition would be a preferred alternative for combating NLRP3-mediated maladies. NLRP3-mediated pyroptosis has been documented by a plethora of contemporary studies as a crucial system adding to the NLRP3 inflammasome linked disease processes [49][50][290,291]. Documentation surfacing has noted gasdermin D (GSDMD) as a controlling protein culpable for pyroptosis [51][52][292,293], making it an enticing therapeutic target for ameliorating NLRP3-evoked pyroptosis associated disorders. Since knowledge of the structure and components of the NLRP3 inflammasome has become available, forthcoming studies should harness this lead and effect progress of development of direct NLRP3 inflammasome inhibitors endowed with increased exactitude and potency. Moreover, nanobodies (Nbs) are lately being scrutinized comprehensively as therapeutics owing to their high precision, stability, and low propensity to induce side effects [53][54][294,295]. It can be foreseen that Nbs may also be assessed for inhibiting NLRP3 inflammasome activation. Significant strides have been undertaken to unravel the NLRP3 inflammasome structure, the processes leading to its activation, and its role in the initiation and evolution of various disorders. Moreover, a multitude of small molecules as NLRP3 inflammasome inhibitors have been documented in the research literature, and a few of these leads have highlighted admirable therapeutic prowess. However, none of them has been approved by Food and Drug Administration (FDA) or other drug regulatory agencies. Contemporary research must continuously concentrate on the development of specific, small-molecule NLRP3 inflammasome activation inhibitors with enhanced pharmacokinetic characteristics, the ability to permeate efficiently across the blood–brain barrier and cell membranes, and it must be affordable too. Undoubtedly, the ongoing contemporary characterization of clinical inflammasome activators and blockers will spur interest in acquiring deeper insights into inflammasome-driven autoinflammatory mechanisms from a futuristic point of view. This is bound to widen therapeutic modalities for patients suffering from NLRP3-mediated metabolic and neurodegenerative diseases as well as certain cancers, where there is a tremendous unmet therapeutic need [55][296]. Finally, an appreciation of the molecular biology in connection with inflammasome priming and activation facilitates the prediction that an array of nutraceuticals could possibly confer salutary clinical promise for impeding inflammasome activity—antioxidants such as phycocyanobilin, phase 2 inducers, melatonin, and N-acetylcysteine, the AMPK activator berberine, glucosamine, zinc, and various nutraceuticals which ramp up production of hydrogen sulfide. Complex nutraceuticals or functional foods consisting of several of such compounds may find value in the obviation and modulation of a great variety of medical disorders [56][297].