Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Christiane Stehmann and Version 2 by Rita Xu.
Genetic prion disease accounts for 10–15% of prion disease. While insertion of four or more octapeptide repeats are clearly pathogenic, smaller repeat insertions have an unclear pathogenicity.
- prion disease
- Creutzfeldt-Jakob disease
- genetic Creutzfeldt-Jakob disease
1. Introduction
Prion disease (PrD) can be sporadic, genetic, or acquired. Genetic prion disease (gPrD) accounts for 10–15% of prion diseases and is caused by pathogenic sequence variations in the prion protein gene (PRNP) [1]. Most PRNP mutations are point mutations but deletions and insertions in the octapeptide repeat region of PRNP are reported, referred to respectively as octapeptide repeat deletions (OPRDs) and octapeptide repeat insertions (OPRIs) [2]. The combination and number of repeats in OPRI mutations vary and are thought to contribute to phenotypic variation [1][3][4][1,3,4]. In this study, we sought to characterize the clinical features, histopathology, and molecular typing of the prion protein (PrP) associated with 2-OPRI patients and to determine the risk for Creutzfeldt–Jakob disease (CJD) in carriers of the 2-OPRI variant.
The first case of the 2-OPRI in the setting of CJD was reported in 1993 [5]. The proband died of neuropathologically verified CJD 3 months after presenting with speech and language difficulties. Multiple first-degree family members were carriers of the 2-OPRI variant, including a family member with a long-standing history of dementia.
A second case of 2-OPRI in the setting of dementia was reported in 2000 [6]. The patient presented after years of progressive memory impairment. Genetic studies revealed a 2-OPRI variant with valine homozygosity at codon 129 of PRNP. No autopsy was performed. The patient did not have a family history suggestive of PrD.
A third case of 2-OPRI in the setting of dementia was reported in 2004 [4]. The patient developed dementia in their sixth decade of life and then declined over years to a mute state. Autopsy results were not reported. Genetic studies demonstrated heterozygosity at codon 129 of PRNP.
2. Clinical Features and Genetics
Eight cases were compiled, and clinical data is shown in Table 1. All cases were of white race. The median age at onset was 75 years (range 58–84 years, mean 71 years ± 8.1). The age at disease onset was known for six cases and mean age at onset differed significantly between methionine homozygotes and heterozygotes at codon 129 (64.3 ± 5.5 and 78.0 ± 5.3, respectively, t-test, p = 0.036). The median disease duration was 7 months (range 2–21 months). All cases with known dates of onset had an illness duration of 7 months or less, with the exception of one case of probable CJD that had an illness duration of 21 months. Family history is relevant because highly penetrant mutations typically show an autosomal dominant inheritance pattern in pedigrees, whereas low penetrance variants might only rarely cause CJD in relatives. Family history of neurodegenerative disease was noted in three cases. No cases had a family history of prion disease. The clinical phenotype of the 2-OPRI cases was heterogenous but marked by typical CJD features, including dementia, ataxia, pyramidal signs, myoclonus, and visual symptoms. One case demonstrated periodic sharp wave complexes on an electroencephalogram. Brain MRIs were obtained in all but one case and were consistent with PrD: all MRIs demonstrated cortical hyperintensity on diffusion weighted imaging (DWI), while three of seven cases demonstrated hyperintensity in the basal ganglia. All but one case demonstrated a positive 14-3-3 in the CSF. The mean CSF tau value was 12,402 pg/mL (high tau levels, e.g., >1500 pg/mL, are suggestive of prion disease) [7][8]. All cases tested by real time quaking-induced conversion (RT-QuIC) were positive. All cases demonstrated two repeats in the octapeptide region of PRNP. Repeat sequences were available in seven cases: Most cases demonstrated repeats in R2-R2 (Table 2). Five cases were homozygous for methionine at codon 129. Three cases were methionine-valine heterozygous.
[8]; resPrPD: PK-resistant disease-associated PrP; Codon 129 polymorphism in cis with the 2-OPRI mutation is underlined if known. US: United States; UK: United Kingdom; AU: Australia; RT-QuIC: real-time quaking-induced conversion; Pos.: positive; Neg.: negative; Unk.: unknown; PSWC: periodic sharp wave complexes; NA: not available; AD: Alzheimer’s disease.
Table 1. Clinical presentation of 2-OPRI cases.
| Case | Origin | Diagnosis | Codon 129 Genotype |
|---|
Table 2. Molecular features and histotype of 2-OPRI cases.
| Case Number | Codon 129 Genotype | Repeat Sequence a | resPrPD Type | resPrPD Type a | Age at Onset (Years) | Disease Duration (Months) | Gender | 14-3-3 Protein | Tau (pg/mL) b | RT-QuIC | PSWC on EEG | MRI c/w CJD | bFamily History | Clinical Phenotype |
|---|
| sCJD Histotype | b | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | US | Definite | MM | 1 | 67 | 2 | Male | Pos. | 17,727 | NA | NA | Yes | None | Slurred speech, then a fulminant course including cognitive and cerebellar symptoms and myoclonus |
| 1 | MM | R1-R2-R2-R2-R2-R3-R4 | 1 | MM(MV)1 | ||||||||||||
| 2 | US | Probable | MM | 1 | NA | NA | Male | Pos. | ||||||||
| 2 | MM | R1-R2-R2-R3-R2-R3-R4 | 8848 | NA | NA | 1 | MM(MV)1 | Yes | None | NA | ||||||
| 3 | US | Definite | MM | 1 | 68 | 7 | Female | Pos. | 7990 | NA | Yes | Yes | Mother with several year slowly progressive dementia in her 60s, thought to be AD | Absence-like episodes, followed 5 months later by cognitive symptoms, personality change, and myoclonus | ||
| 4 | US | Probable | R2-R2MV | 1–2 | 84 | 3 | Female | Unk. | Unk. | NA | NA | Yes | None | Myoclonus, unilateral weakness/spasticity, late cognitive symptoms | ||
| -R3-R4 | 1-2 | MV2C + 1 | c | 5 | US | Definite | MM | 1–2 | NA | NA | Female | Pos. | 15,046 | Pos. | NA | |
| 5 | MM | R1-R2-R2-R3-R2a-R2a-R4 | 1 | MM1-2UK | Definite | MM | 1 | 58 | 7 | Male | Unk. | Unk. | NA | No | ||
| 3 | MM | R1-R2-R2-R2-R2-R3-R4 | 1 | MM(MV)1 | ||||||||||||
| 4 | MV | R1-R2-R2- | Yes | c | None | NA | ||||||||||
| 6 | UK | Probable | MV | NA | 76 | 10 | Male | Neg. | Unk. | Pos. | No | Yes | Sister with 2-year history of Alzheimer’s dementia starting at age 89; sister with mild dementia in her 80s. | Gait ataxia, followed by cognitive symptoms, visual hallucinations, and myoclonus | ||
| 6 (UK) | MV | R1-R2-R2-R2-R2-R3-R4 | NA | NA | 7 | NA | ||||||||||
| 7 (UK) | MM | R1-R2-R2-R3-R2a-R2a-R4 | Unknown | Right hand paresthesia followed by unilateral weakness/spasticity | ||||||||||||
| 1 | MM(MV)1. | 8 | AU | Probable | MV | NA | 74 | 21 | Female | Pos. | Unk. | Pos. | No | Yes | Brother with 5-year history of dementia in his 60s. | Visual disturbances, followed by gait disturbance, Parkinsonian features, apraxia, and visuospatial difficulties |
a As defined by Goldfarb et al. [9][14]; b Parchi et al. classification [8][24]; c Minor sCJDMM2 component affecting the temporal cortex; Codon 129 polymorphism in cis with the 2-OPRI mutation is underlined if known. resPrPD: PK-resistant disease-associated PrP; MM: methionine homozygosity; MV: methionine/valine heterozygosity; NA: not available.
3. Histopathology and Immunohistochemistry
Neuropathology was available in six out of eight cases. Four out of five cases demonstrated classic spongiform degeneration (SD) and reactive astrocytosis on hematoxylin and eosin staining with a synaptic pattern of PrP deposition on immunohistochemical examination (cases 1–3 and 7, Table 2) (Figure 1). These histopathological features matched those associated with sCJDMM(MV)1. case 5 demonstrated the sCJDMM1-2 histotype with a minor type 2 component, characterized by large vacuoles and perivacuolar PrP deposition in the temporal cortex. case 4, carrying the 129MV genotype and resPrPD type 1–2, showed mild SD with small and large vacuoles, and diffuse and coarse PrP deposits within the neocortex. No features suggestive of gPrD, such as filamentous PrP deposits in the subcortical white matter or “striped” cerebellar PrP staining characteristic of some OPRIs, were observed in the autopsied brains [10][11][27,28].
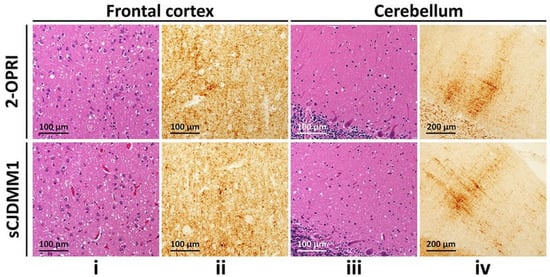
Figure 1. Histological determination of 2-OPRI and sCJDMM1: (i,iii): Hematoxylin-eosin staining. (ii,iv): PrP immunohistochemistry. (i,iii): Fine spongiform degeneration. (ii,iv): Diffuse PrP immunostaining (ii,iv) with the typical “brush stroke-like” deposits in the cerebellar molecular layer (iv); antibody: 3F4.
4. Characterization of Detergent-Insoluble and PK-Resistant PrPD (resPrPD)
Total PrP of the 2-OPRI (cases 2–4) showed several fragments spanning from ~26 to ~35 kDa. Thus, the PrP profile of 2-OPRI differed markedly from that of sCJD, which consisted of three major bands corresponding to di- (~33 kDa), mono- (~31 kDa), and un-glycosylated (~27 kDa) PrP isoforms (Figure 2A). Following high-speed centrifugation, we generated detergent-insoluble PrPD. Western blot analysis demonstrated that most of the PrP fragments of the 2-OPRI cases are detergent-insoluble and that most of them (e.g., 29, 30, 32 kDa, and a doublet of ~34–35 kDa) are not detectable in sCJD. (Figure 2B). Furthermore, the 2-OPRI PrPD profile was different from that observed in a 7-OPRI case. Notably, the ~34–35 kDa doublet appeared as a triplet in the 7-OPRI case, while a band with the molecular weight of ~42–44 kDa was not detected in 2-OPRI (Figure 2C) [12][18]. Following digestion with PK, the Western blot profile of resPrPD from the OPRI and sCJD cases became virtually indistinguishable [12][13][14][18,29,30].
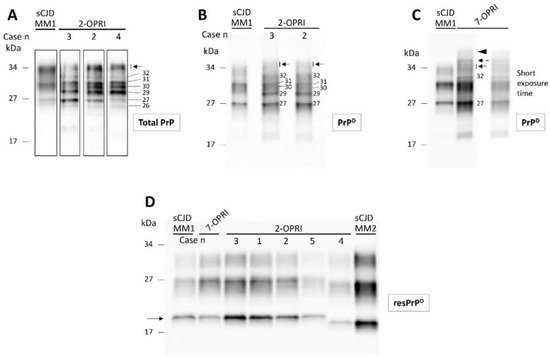
Figure 2. Western blot profiles of total PrP, detergent-insoluble PrPD and resPrPD: Samples harvested from the cerebral cortex were probed with the anti-PrP antibody 3F4. (A): Total PrP showing a complex PrP profile in 2-OPRI cases but not in sCJDMM1. The approximate molecular size of each band is indicated by the numbers on the right; arrow: PrP doublet of ~34–35 kDa. (B): Western blot (WB) profile of detergent-insoluble PrPD harvested from 2-OPRI resembles that of total PrP except for the absence and weak presence of the ~26 and ~30 kDa fragments, respectively. (C): Insoluble PrPD WB profile of a 7-OPRI features a sharp band of ~32 kDa, a PrPD triplet in the ~34–36 kDa region, and a PrPD smear of ~42–44 kDa, whereas the PrPD region of ~31 to 27 kDa resembles that of sCJDMM1. (D): The un-glycosylated isoform of resPrPD (arrow) in 2-OPRI-MM1 (cases 1–3), 2-OPRI-MM1-2 (case 5), and 7-OPRI-MM1 migrates to ~20 kDa, matching the gel mobility of resPrPD type 1 (sCJDMM1). The un-glycosylated resPrPD of 2-OPRI-MV1-2 (case 4) migrates to ~19 kDa, thus matching the gel mobility of sCJDMM2 resPrPD type 2.
5. Estimation of 2-OPRI Penetrance
Interrogation of a number of available large-scale population genetic datasets identified a total of sixteen 2-OPRI alleles in presumably unaffected individuals. Within these 16 alleles, 6 (5 exomic, 1 genomic) were from gnomAD v2.1.1 (USA), 8 from gnomAD v3 (USA), and 2 from the 100,000 Genomes Project (UK) [15][31]. Of the 14 alleles from the gnomAD datasets, 7 were non-Finnish Europeans, 4 were Finnish Europeans, 1 was South Asian, and 1 was African and 1 Latino/admixed American; 8 were male and 6 were female. Age range data was available for 5 of 6 gnomAD v2.1.1 cases only, with 2 cases in the 55–60 years range, 2 cases in the 60–65 year range, 1 case in the 65–70 years age range, and unknown for 1; age range data for gnomAD v3.1.1 is available for 2 of 8 cases, with 1 case in the 65-70 years age range and 1 case in the 70-75 years age range. Estimation of 2-OPRI penetrance and 95% CI using a Bayesian approach and Wilson interval, respectively, revealed an extremely low penetrance of 0.3% and below, and upper bounds of 95% CI below 2% [16][25]. Specifically, the estimated penetrance by leveraging the gnomAD v2.1.1 was 0.34% (95% CI 0.08, 1.46), gnomAD v3 was 0.13% (95%CI 0.03, 0.51), and, for the 100,000 Genomes Project, it was 0.24% (95%CI 0.03, 1.75).