Sepsis remains a common cause of death in intensive care units, accounting for approximately 20% of total deaths worldwide. Its pathogenesis is partly attributable to dysregulated inflammatory responses to bacterial endotoxins (such as lipopolysaccharide, LPS), which stimulate innate immune cells to sequentially release early cytokines (such as tumor necrosis factor (TNF) and interferons (IFNs)) and late mediators (such as high-mobility group box 1, HMGB1). Below is a brief summary of the intricate mechanisms underlying the regulation of bacterial endotoxin-induced HMGB1 release.
- sepsis
- pyroptosis
- innate immune cells
1. Introduction
Microbial infections and resultant sepsis syndromes are the most common causes of death in intensive care units, accounting for approximately 20% of total deaths worldwide
. The pathogenesis of sepsis remains poorly understood, but is partly attributable to immune over-activation or immunosuppression propagated by dysregulated innate immune responses to lethal infections
. Innate immune cells (such as macrophages, monocytes and neutrophils) constitute a front line of defense against microbial infections by eliminating invading pathogens via phagocytosis, and initiating inflammatory responses via various mediators. Upon detection of microbial products such as bacterial endotoxins (lipopolysaccharide, LPS), circulating neutrophils and monocytes immediately infiltrate into infected tissues
. After engulfing and killing pathogens, neutrophils exhaust intracellular enzymes and undergo apoptotic cell death. The cell debris of these apoptotic neutrophils are then removed by tissue macrophages (e.g., Kupffer cells, dendritic cells, and glia cells)
terminally differentiated from infiltrated monocytes.
Innate immune cells also carry various pattern recognition receptors (PRRs) to recognize distinct classes of molecules shared by a group of related microbes, which are collectively termed “pathogen-associated molecular pattern molecules” (PAMPs). For instance, Toll-like Receptor 2 (TLR2)
, TLR4
and TLR9
, respectively, serve as PRRs for distinct PAMPs such as peptidoglycans, bacterial endotoxins, and microbial un-methylated CpG-DNAs. The engagement of various PRRs by different PAMPs similarly activates innate immune cells to sequentially release early cytokines (such as tumor necrosis factor (TNF) and interferons (IFNs)) and late-acting pro-inflammatory mediators [such as high-mobility group box 1 (HMGB1)
and sequestosome 1 (SQSTM1)]
.
HMGB1 is constitutively expressed by most types of cells to maintain a large “pool” of preformed protein in the nucleus, possibly due to the presence of two lysine-rich nuclear localization sequences (NLS)
. It carries two internal repeats of positively charged domains (“HMG boxes” known as “A box” and “B box”) in the N-terminus, and a continuous stretch of negatively charged (aspartic and glutamic acid) residues in the C-terminus. These HMG boxes enable HMGB1 to bind chromosomal DNA, and fulfill its nuclear functions to maintain nucleosomal structure and stability, and regulate gene expression
. Once released, extracellular HMGB1 can bind many endogenous proteins, thereby modulating divergent innate immune responses to lethal infections
.
2. Role of Cytoplasmic PRRs (Caspase-11/4/5/1) in the Regulation of Pyroptosis and HMGB1 Release
We and others demonstrated that ultra-pure LPS (free from any contaminating bacterial proteins, lipids, or nucleic acids) completely failed to induce HMGB1 release, unless the initial LPS priming was accompanied by a second stimulus (e.g., ATP)
. However, crude LPS that might carry trace amounts of bacterial proteins, lipids and nucleic acids, triggered a marked HMGB1 release
. It is possible that some contaminating bacterial proteins and lipids might enhance endocytosis of LPS, and consequently facilitate its innate recognition by cytoplasmic PRRs such as Casp-11/4/5. Indeed, when LPS was delivered to cytoplasmic Casp-11/4/5 either via CD14/TLR4 receptor-mediated endocytosis or bacteria-derived outer membrane vesicles (OMV)
, it induced “non-canonical” inflammasome activation via oligomerization and proximity-induced activation of Casp-11/4/5 (
)
. The activated Casp-11/4/5 then catalyzes the cleavage of Gasdermin D (GSDMD) to form cytoplasmic membrane pores that cause immediate ionic gradient loss, osmotic burst and cell membrane rupture, a process aforementioned as “pyroptosis”. For the optimal activation of non-canonical inflammasome, both type I IFN-α/β and type II IFN-γ are needed to up-regulate Casp-11/4/5
as well as guanylate-binding proteins
responsible for disrupting pathogen-containing vacuoles and releasing LPS. Coincidently, we and others demonstrated that LPS-inducible type I IFN-α/β
and type II IFN-γ
effectively stimulated innate immune cells to release HMGB1 in a time- and dose-dependent fashion.
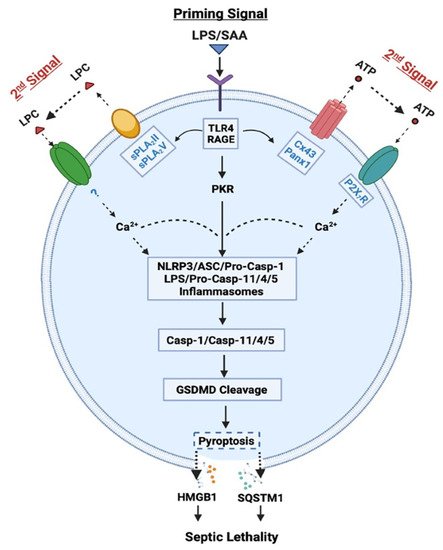
Role of Casp-1-mediated canonical and Casp-11/4/5-mediated non-canonical inflammasome activation in LPS- or SAA-induced pyroptosis and HMGB1 release. LPS or SAA may prime innate immune cells to up-regulate the expression of Cx43/Panx1 hemichannels, sPLA
s and interferon-induced double-stranded RNA-activated protein kinase (PKR), thereby eliciting the release of ATP or LPC that may activate P2X
R- or other receptor-mediated Ca
signaling. It then induces a feed-forwarding activation of PKR and inflammasome, cleavage of GSDMD, pyroptosis, and subsequent release of late mediators (such as HMGB1 and SQSTM1) of lethal infections.
In contrast, the “canonical” inflammasome activation is characterized by the oligomerization of intracellular “nucleotide-binding oligomerization domain (NOD)-like receptors” (NLRs such as NLRP1, NLRP3, and NLRC4) and the “apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain” (ASC) adaptor, as well as the recruitment and activation of pro-Casp-1 (
)
. Specifically, the pro-Casp-1 forms a heteromeric protein complex with an ASC adaptor and a NLR receptor, and the resultant protein complex, termed the “inflammasome”, is responsible for cleaving pro-Casp-1 to generate Casp-1, which triggers canonical inflammasome activation and pyroptosis via GSDMD cleavage
. Likewise, the optimal activation of canonical inflammasome also depends on a two-step process: (1) a priming signal elicited by extracellular PAMPs (e.g., LPS) to up-regulate NLRP3 expression; and (2) a secondary signal elicited by extracellular damage-associated molecular pattern (DAMPs, e.g., ATP) to induce NLRP3 oligomerization with ASC and pro-Casp-1 (
). Notably, the cleavage of pannexin-1 (Panx1) hemichannel by Casp-11/4/5 might be needed for releasing ATP and activating the purinergic P2X
receptor (P2X
R) and inflammasome signalings (
)
. Consistently, we found that crude LPS also markedly up-regulated Panx1 expression in macrophages and monocytes, and consequently elevated their hemichannel activities to release ATP
, supporting a pathogenic role of Panx1 in LPS-induced HMGB1 release and animal lethality
(
).
It is thus possible that following cytoplasmic translocation, HMGB1 could be secreted extracellularly through Casp-1- or Casp-11/4/5-mediated inflammasome activation and pyroptosis (
). Recent evidence suggested that inflammasome-dependent HMGB1 release could not occur immediately after the formation of GSDMD membrane pores, but became prominent following the rupture of cytoplasmic membranes
. Consistently, pharmacological inhibition (with a broad-spectrum Caspase inhibitor Z-VAD-FMK) or genetic disruption of key inflammasome components (e.g., Casp-1 or Nlrp3) uniformly blocked the LPS/ATP-induced HMGB1 secretion
. Likewise, genetic disruption of interferon-induced double-stranded RNA-activated protein kinase (PKR) expression or pharmacological inhibition of its phosphorylation similarly reduced the LPS-induced inflammasome activation
, pyroptosis
, and HMGB1 release
. Thus, crude LPS may prime macrophages by simultaneously up-regulating PKR expression and eliciting Panx-1-mediated ATP release, thereby activating P2X
R
to induce a feed-forwarding PKR/inflammasome activation, pyroptosis and HMGB1 secretion (
).
In addition, HMGB1 can also be passively released by somatic cells undergoing cytoplasmic membrane destruction due to accidental mechanical events or regulated processes governed by other caspases or kinases. For instance, circulating levels of HMGB1 were rapidly elevated in critical ill patients with non-penetrating trauma
, thereby contributing to trauma-induced dysregulated inflammation, immune paralysis or immunosuppression. Even following viral infections with influenza
or SARS-CoV-2
, proinflammatory cytokines such as TNF and IFN-γ can also induce necroptosis
or PANoptosis
via other caspases and kinases such as the Receptor-Interacting Serine/Threonine Kinase 3 (RIPK3)
and Casp-8
. Thus, various cell death pathways can potentially lead to the passive release of HMGB1 following traumatic injuries or microbial infections. However, the possible roles of HMGB1 and various other cytokines in the pathogenesis of lethal infections such as COVID-19 remain controversial, because there is still a lack of clear association between many cytokine biomarkers and the severity of viral infections
.
3. Pathogenic Role of Extracellular HMGB1 in Dysregulated Inflammation, Immunosuppression, and Immune Paralysis
Once released, extracellular HMGB1 can bind various PRRs and PAMPs to orchestrate divergent inflammatory responses. For instance, HMGB1 can bind TLR4
, TLR9
, receptor for advanced glycation end products (RAGE)
, cluster of differentiation 24 (CD24)/Siglec-10
, Mac-1
, or single-transmembrane-domain proteins (e.g., syndecans)
. Due to its relatively higher affinity to TLR4 (K
= 22.0 nM)
and lower affinity to RAGE (K
= 97.7-710 nM)
, HMGB1 might first bind TLR4 when it was actively secreted by innate immune cells at relatively lower amounts
. Consequently, it could directly activate macrophages
, neutrophils
and endothelial cells
to produce various cytokines and chemokines
partly through MyD88-IRAK4-dependent signaling pathways (
A).
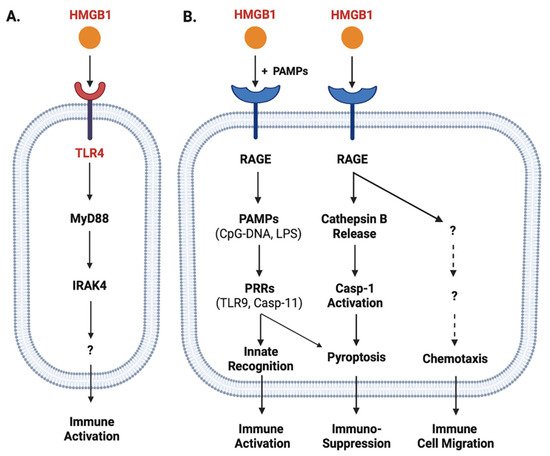
Role of TLR4 and RAGE in the regulation of HMGB1-mediated divergent inflammatory responses. HMGB1 can bind different PRRs such as TLR4 (Panel
) and RAGE (Panel
) with different affinities, and consequently induce divergent inflammatory responses such as immune cell migration, immune activation, or pyroptosis and resultant immunosuppression.
When HMGB1 was passively released by innate immune and somatic cells at relatively higher levels, it might also bind various microbial PAMPs (e.g., CpG-DNA or LPS) and RAGE
and consequently promoted RAGE-receptor-mediated endocytosis of these microbial products (
B)
. Upon reaching acidic endosomal and lysosomal compartments near HMGB1′s isoelectric pH, HMGB1 became neutrally charged and set free its cargos (LPS or CpG-DNA)
, thereby facilitating their recognition by respective PRRs such as TLR9
or Casp-11
to augment inflammatory responses (
B). Furthermore, the engagement of RAGE with HMGB1 might also induce chemotaxis
and the migration of monocytes, dendritic cells
and neutrophils
, thereby facilitating the recruitment of innate immune cells to site of the infection to orchestrate inflammatory responses
(
B). Finally, the engagement of HMGB1 with RAGE
might also induce TLR4 internalization and desensitization to subsequent stimulus (e.g., endotoxin), and might even trigger macrophage pyroptosis
via a cascade of events including cathepsin B release from ruptured lysosomes followed by pyroptosome formation and Casp-1 activation (
B).
In neutrophils, HMGB1 can bind TLR4 to promote the formation of neutrophil extracellular traps (NETs), thereby amplifying neutrophil-mediated inflammatory responses
. In contrast, the engagement of RAGE by HMGB1 can adversely impair neutrophil NADPH-dependent production of reactive oxidation species (ROS) and associated bacterial killing, contributing to sepsis-induced immune paralysis and immuno-suppression
. Consistently, the blockade of extracellular HMGB1 activities with neutralizing antibodies even during a late stage of sepsis still restored neutrophil NADPH activity and anti-bacterial capacities
. Thus, excessive HMGB1 release contributes to the pathogenesis of lethal infections by posing divergent adverse effects such as immune tolerance
, immune paralysis
and immunosuppression
(
B).
4. Positive Regulators of LPS-Induced HMGB1 Release
In addition to LPS-inducible type I IFN-α/β
and type II IFN-γ
, human serum amyloid A (SAA) also effectively induced HMGB1 release by innate immune cells in a TLR4/RAGE-dependent fashion
(
). Consistent with its capacity in stimulating NLRP3 inflammasome activation
, we observed that SAA also stimulated PKR expression and phosphorylation
. Conversely, pharmacological inhibition of PKR inhibited SAA-induced HMGB1 release
, supporting an important role for PKR phosphorylation, inflammasome activation and pyroptosis in the SAA-induced HMGB1 release (
). In addition, some LPS-inducible enzymes [such as the 14 kDa type II secretory phospholipase A
(sPLA
), inducible nitric oxide synthase (iNOS), and pyruvate kinase M2 (PKM2)] were also implicated in the regulation of LPS-induced HMGB1 release (
)
. In agreement with these findings, we found that human SAA effectively up-regulated the expression of sPLA
-IIE and sPLA
-V in murine macrophages (
and
)
, and concurrently induced HMGB1 release
. Conversely, the suppression of sPLA
-IIE expression by high density lipoproteins (HDL) also attenuated SAA-induced HMGB1 release, supporting a role of sPLA
in the regulation of HMGB1 release
. It is not yet known whether sPLA
s facilitate HMGB1 release partly by catalyzing the production of lyso-phosphatidylcholine (LPC) and leukotrienes that are capable of activating NLRP3 inflammasome and pyroptosis (
)
.
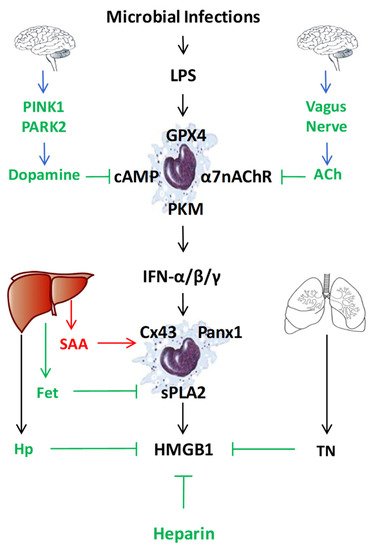
Endogenous regulators of LPS-induced HMGB1 release or action. To regulate the LPS-induced HMGB1 release or action, mammals have evolved multiple regulatory mechanisms that include neuro-immune pathways, liver-derived acute-phase proteins (e.g., SAA, Fetuin-A (Fet), Haptoglobin (Hp)), as well as other endogenous proteins (e.g., tetranectin (TN)) or polysaccharides (heparin).
Finally, both crude LPS and human SAA effectively up-regulated the expression of hemichannel molecules such as Panx1
and Connexin 43 (Cx43)
in innate immune cells (
and
). The possible role of Cx43 in the regulation of LPS-induced HMGB1 release was supported by our findings that several Cx43 mimetic peptides, the GAP26 and Peptide 5 (ENVCYD), simultaneously attenuated LPS-induced hemichannel activation and HMGB1 release
. It was further supported by observation that genetic disruption of macrophage-specific Cx43 expression conferred protection against lethal endotoxemia and sepsis
. It is possible that Cx43 hemichannel provides a temporal mode of ATP release
, which then contributes to the LPS-stimulated PKR phosphorylation, inflammasome activation, pyroptosis and HMGB1 secretion (
and
)
. Intriguingly, recent evidence has suggested that macrophages also form Cx43-containing gap junction with non-immune cells such as cardiomyocytes
, epithelial
and endothelial cells
. It is possible that innate immune cells may communicate with non-immune cells through Cx43-containing gap junction channels to regulate HMGB1 release and to orchestrate inflammatory responses
. Interestingly, recent studies have revealed an important role of lipid peroxidation
and cAMP immune-metabolism
in the regulation of Casp-11-mediated “non-canonical” inflammasome activation and pyroptosis (
). However, the possible role of these immunometabolism pathways in the regulation of LPS-induced HMGB1 release remains an exciting subject of future investigations.
References
- Rudd, K.E.; Johnson, S.C.; Agesa, K.M.; Shackelford, K.A.; Tsoi, D.; Kievlan, D.R.; Colombara, D.V.; Ikuta, K.S.; Kissoon, N.; Finfer, S.; et al. Global, regional, and national sepsis incidence and mortality, 1990-2017: Analysis for the Global Burden of Disease Study. Lancet 2020, 395, 200–211. [Google Scholar] [CrossRef]
- Hotchkiss, R.S.; Coopersmith, C.M.; McDunn, J.E.; Ferguson, T.A. The sepsis seesaw: Tilting toward immunosuppression. Nat. Med. 2009, 15, 496–497. [Google Scholar] [CrossRef]
- Tang, D.; Wang, H.; Billiar, T.R.; Kroemer, G.; Kang, R. Emerging mechanisms of immunocoagulation in sepsis and septic shock. Trends. Immunol. 2021, 42, 508–522. [Google Scholar] [CrossRef] [PubMed]
- Luster, A.D.; Alon, R.; von Andrian, U.H. Immune cell migration in inflammation: Present and future therapeutic targets. Nat. Immunol. 2005, 6, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Liles, W.C. Immunomodulatory approaches to augment phagocyte-mediated host defense for treatment of infectious diseases. Semin. Respir. Infect. 2001, 16, 11–17. [Google Scholar] [CrossRef]
- Brightbill, H.D.; Libraty, D.H.; Krutzik, S.R.; Yang, R.B.; Belisle, J.T.; Bleharski, J.R.; Maitland, M.; Norgard, M.V.; Plevy, S.E.; Smale, S.T.; et al. Host defense mechanisms triggered by microbial lipoproteins through toll-like receptors. Science 1999, 285, 732–736. [Google Scholar] [CrossRef]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.Y.; Huffel, C.V.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef] [PubMed]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar] [CrossRef]
- Wang, H.; Bloom, O.; Zhang, M.; Vishnubhakat, J.M.; Ombrellino, M.; Che, J.; Frazier, A.; Yang, H.; Ivanova, S.; Borovikova, L.; et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science 1999, 285, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Liu, J.; Zeng, L.; Zhu, S.; Wang, H.; Billiar, T.R.; Kroemer, G.; Klionsky, D.J.; Zeh, H.J.; Jiang, J.; et al. Extracellular SQSTM1 mediates bacterial septic death in mice through insulin receptor signalling. Nat. Microbiol. 2020, 5, 1576–1587. [Google Scholar] [CrossRef]
- Bonaldi, T.; Talamo, F.; Scaffidi, P.; Ferrera, D.; Porto, A.; Bachi, A.; Rubartelli, A.; Agresti, A.; Bianchi, M.E. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003, 22, 5551–5560. [Google Scholar] [CrossRef] [PubMed]
- Bustin, M. At the crossroads of necrosis and apoptosis: Signaling to multiple cellular targets by HMGB1. Sci. STKE. 2002, 2002, E39. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Chen, R.; Zhang, Q.; Hou, W.; Wu, S.; Cao, L.; Huang, J.; Yu, Y.; Fan, X.G.; Yan, Z.; et al. HMGB1 in health and disease. Mol. Aspects. Med. 2014, 40, 1–116. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Sarkar, A.; Vande, W.L.; Vitari, A.C.; Amer, A.O.; Wewers, M.D.; Tracey, K.J.; Kanneganti, T.D.; Dixit, V.M. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J. Immunol. 2010, 185, 4385–4392. [Google Scholar] [CrossRef]
- Lu, B.; Nakamura, T.; Inouye, K.; Li, J.; Tang, Y.; Lundback, P.; Valdes-Ferrer, S.I.; Olofsson, P.S.; Kalb, T.; Roth, J.; et al. Novel role of PKR in inflammasome activation and HMGB1 release. Nature 2012, 488, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Vanaja, S.K.; Russo, A.J.; Behl, B.; Banerjee, I.; Yankova, M.; Deshmukh, S.D.; Rathinam, V.A.K. Bacterial Outer Membrane Vesicles Mediate Cytosolic Localization of LPS and Caspase-11 Activation. Cell 2016, 165, 1106–1119. [Google Scholar] [CrossRef] [PubMed]
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS activates caspase-11: Implications in TLR4-independent endotoxic shock. Science 2013, 341, 1250–1253. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.; Vanaja, S.K.; Waggoner, L.; Sokolovska, A.; Becker, C.; Stuart, L.M.; Leong, J.M.; Fitzgerald, K.A. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell 2012, 150, 606–619. [Google Scholar] [CrossRef]
- Aachoui, Y.; Kajiwara, Y.; Leaf, I.A.; Mao, D.; Ting, J.P.; Coers, J.; Aderem, A.; Buxbaum, J.D.; Miao, E.A. Canonical Inflammasomes Drive IFN-γ to Prime Caspase-11 in Defense against a Cytosol-Invasive Bacterium. Cell. Host. Microbe. 2015, 18, 320–332. [Google Scholar] [CrossRef]
- Meunier, E.; Dick, M.S.; Dreier, R.F.; SchÃrmann, N.; Kenzelmann, B.D.; Warming, S.; Roose-Girma, M.; Bumann, D.; Kayagaki, N.; Takeda, K.; et al. Caspase-11 activation requires lysis of pathogen-containing vacuoles by IFN-induced GTPases. Nature 2014, 509, 366–370. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, S.J.; Lee, I.S.; Lee, M.S.; Uematsu, S.; Akira, S.; Oh, K.I. Bacterial endotoxin induces the release of high mobility group box 1 via the IFN-beta signaling pathway. J. Immunol. 2009, 182, 2458–2466. [Google Scholar] [CrossRef]
- Yang, X.; Cheng, X.; Tang, Y.; Qiu, X.; Wang, Z.; Fu, G.; Wu, J.; Kang, H.; Wang, J.; Wang, H.; et al. The role of type 1 interferons in coagulation induced by gram-negative bacteria. Blood 2020, 135, 1087–1100. [Google Scholar] [CrossRef]
- Rendon-Mitchell, B.; Ochani, M.; Li, J.; Han, J.; Wang, H.; Yang, H.; Susarla, S.; Czura, C.; Mitchell, R.A.; Chen, G.; et al. IFN-gamma Induces High Mobility Group Box 1 Protein Release Partly Through a TNF-Dependent Mechanism. J. Immunol. 2003, 170, 3890–3897. [Google Scholar] [CrossRef]
- Yang, D.; He, Y.; Munoz-Planillo, R.; Liu, Q.; Nunez, G. Caspase-11 Requires the Pannexin-1 Channel and the Purinergic P2X7 Pore to Mediate Pyroptosis and Endotoxic Shock. Immunity 2015, 43, 923–932. [Google Scholar] [CrossRef]
- Chiu, Y.H.; Jin, X.; Medina, C.B.; Leonhardt, S.A.; Kiessling, V.; Bennett, B.C.; Shu, S.; Tamm, L.K.; Yeager, M.; Ravichandran, K.S.; et al. A quantized mechanism for activation of pannexin channels. Nat. Commun. 2017, 8, 14324. [Google Scholar] [CrossRef]
- Chen, W.; Zhu, S.; Wang, Y.; Li, J.; Qiang, X.; Zhao, X.; Yang, H.; D’Angelo, J.; Becker, L.; Wang, P.; et al. Enhanced Macrophage Pannexin 1 Expression and Hemichannel Activation Exacerbates Lethal Experimental Sepsis. Sci. Rep. 2019, 9, 160–37232. [Google Scholar] [CrossRef] [PubMed]
- Volchuk, A.; Ye, A.; Chi, L.; Steinberg, B.E.; Goldenberg, N.M. Indirect regulation of HMGB1 release by gasdermin D. Nat. Commun. 2020, 11, 4561–18443. [Google Scholar] [CrossRef]
- Phulphagar, K.; Kühn, L.I.; Ebner, S.; Frauenstein, A.; Swietlik, J.J.; Rieckmann, J.; Meissner, F. Proteomics reveals distinct mechanisms regulating the release of cytokines and alarmins during pyroptosis. Cell. Rep. 2021, 34, 108826. [Google Scholar] [CrossRef]
- Qin, S.; Wang, H.; Yuan, R.; Li, H.; Ochani, M.; Ochani, K.; Rosas-Ballina, M.; Czura, C.J.; Huston, J.M.; Miller, E.; et al. Role of HMGB1 in apoptosis-mediated sepsis lethality. J. Exp. Med. 2006, 203, 1637–1642. [Google Scholar] [CrossRef] [PubMed]
- Hett, E.C.; Slater, L.H.; Mark, K.G.; Kawate, T.; Monks, B.G.; Stutz, A.; Latz, E.; Hung, D.T. Chemical genetics reveals a kinase-independent role for protein kinase R in pyroptosis. Nat. Chem. Biol. 2013, 9, 398–405. [Google Scholar] [CrossRef]
- Surprenant, A.; Rassendren, F.; Kawashima, E.; North, R.A.; Buell, G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 1996, 272, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.J.; Brohi, K.; Calfee, C.S.; Rahn, P.; Chesebro, B.B.; Christiaans, S.C.; Carles, M.; Howard, M.; Pittet, J.F. Early release of high mobility group box nuclear protein 1 after severe trauma in humans: Role of injury severity and tissue hypoperfusion. Crit. Care. 2009, 13, R174. [Google Scholar] [CrossRef]
- Peltz, E.D.; Moore, E.E.; Eckels, P.C.; Damle, S.S.; Tsuruta, Y.; Johnson, J.L.; Sauaia, A.; Silliman, C.C.; Banerjee, A.; Abraham, E. HMGB1 is markedly elevated within 6 hours of mechanical trauma in humans. Shock 2009, 32, 17–22. [Google Scholar] [CrossRef]
- Huang, L.F.; Yao, Y.M.; Dong, N.; Yu, Y.; He, L.X.; Sheng, Z.Y. Association of high mobility group box-1 protein levels with sepsis and outcome of severely burned patients. Cytokine 2011, 53, 29–34. [Google Scholar] [CrossRef]
- Zhang, T.; Yin, C.; Boyd, D.F.; Quarato, G.; Ingram, J.P.; Shubina, M.; Ragan, K.B.; Ishizuka, T.; Crawford, J.C.; Tummers, B.; et al. Influenza Virus Z-RNAs Induce ZBP1-Mediated Necroptosis. Cell 2020, 180, 1115–1129. [Google Scholar] [CrossRef]
- Nogusa, S.; Thapa, R.J.; Dillon, C.P.; Liedmann, S.; Oguin, T.H., III; Ingram, J.P.; Rodriguez, D.A.; Kosoff, R.; Sharma, S.; Sturm, O.; et al. RIPK3 Activates Parallel Pathways of MLKL-Driven Necroptosis and FADD-Mediated Apoptosis to Protect against Influenza A Virus. Cell Host. Microbe 2016, 20, 13–24. [Google Scholar] [CrossRef]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-α and IFN-γ Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2021, 184, 149–168. [Google Scholar] [CrossRef]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Thapa, R.J.; Nogusa, S.; Chen, P.; Maki, J.L.; Lerro, A.; Andrake, M.; Rall, G.F.; Degterev, A.; Balachandran, S. Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proc. Natl. Acad. Sci. USA 2013, 110, E3109–E3118. [Google Scholar] [CrossRef] [PubMed]
- Gunther, C.; Martini, E.; Wittkopf, N.; Amann, K.; Weigmann, B.; Neumann, H.; Waldner, M.J.; Hedrick, S.M.; Tenzer, S.; Neurath, M.F.; et al. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature 2011, 477, 335–339. [Google Scholar] [CrossRef]
- Mehta, P.; Fajgenbaum, D.C. Is severe COVID-19 a cytokine storm syndrome: A hyperinflammatory debate. Curr. Opin. Rheumatol. 2021, 33, 419–430. [Google Scholar] [CrossRef]
- Sinha, P.; Matthay, M.A.; Calfee, C.S. Is a “Cytokine Storm” Relevant to COVID-19? JAMA. Intern. Med. 2020, 180, 1152–1154. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Wang, H.; Ding, A.; Golenbock, D.T.; Latz, E.; Czura, C.J.; Fenton, M.J.; Tracey, K.J.; Yang, H. HMGB1 signals through Toll-like Receptor (TLR) 4 and TLR2. Shock 2006, 26, 174–179. [Google Scholar] [CrossRef]
- Ha, T.; Xia, Y.; Liu, X.; Lu, C.; Liu, L.; Kelley, J.; Kalbfleisch, J.; Kao, R.L.; Williams, D.L.; Li, C. Glucan phosphate attenuates myocardial HMGB1 translocation in severe sepsis through inhibiting NF-kappaB activation. Am. J. Physiol Heart Circ. Physiol. 2011, 301, H848–H855. [Google Scholar] [CrossRef] [PubMed]
- Xiang, M.; Shi, X.; Li, Y.; Xu, J.; Yin, L.; Xiao, G.; Scott, M.J.; Billiar, T.R.; Wilson, M.A.; Fan, J. Hemorrhagic shock activation of NLRP3 inflammasome in lung endothelial cells. J. Immunol. 2011, 187, 4809–4817. [Google Scholar] [CrossRef]
- Ivanov, S.; Dragoi, A.M.; Wang, X.; Dallacosta, C.; Louten, J.; Musco, G.; Sitia, G.; Yap, G.S.; Wan, Y.; Biron, C.A.; et al. A novel role for HMGB1 in TLR9-mediated inflammatory responses to CpG-DNA. Blood 2007, 110, 1970–1981. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Avalos, A.M.; Mao, S.Y.; Chen, B.; Senthil, K.; Wu, H.; Parroche, P.; Drabic, S.; Golenbock, D.; Sirois, C.; et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol. 2007, 8, 487–496. [Google Scholar] [CrossRef]
- Chen, G.Y.; Tang, J.; Zheng, P.; Liu, Y. CD24 and Siglec-10 selectively repress tissue damage-induced immune responses. Science 2009, 323, 1722–1725. [Google Scholar] [CrossRef] [PubMed]
- Orlova, V.V.; Choi, E.Y.; Xie, C.; Chavakis, E.; Bierhaus, A.; Ihanus, E.; Ballantyne, C.M.; Gahmberg, C.G.; Bianchi, M.E.; Nawroth, P.P.; et al. A novel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1-integrin. EMBO J. 2007, 26, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Salmivirta, M.; Rauvala, H.; Elenius, K.; Jalkanen, M. Neurite growth-promoting protein (amphoterin, p30) binds syndecan. Exp. Cell. Res. 1992, 200, 444–451. [Google Scholar] [CrossRef]
- Yang, H.; Hreggvidsdottir, H.S.; Palmblad, K.; Wang, H.; Ochani, M.; Li, J.; Lu, B.; Chavan, S.; Rosas-Ballina, M.; Al Abed, Y.; et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc. Natl. Acad. Sci. USA 2010, 107, 11942–11947. [Google Scholar] [CrossRef] [PubMed]
- Ling, Y.; Yang, Z.Y.; Yin, T.; Li, L.; Yuan, W.W.; Wu, H.S.; Wang, C.Y. Heparin changes the conformation of high-mobility group protein 1 and decreases its affinity toward receptor for advanced glycation endproducts in vitro. Int. Immunopharmacol. 2011, 11, 187–193. [Google Scholar] [CrossRef]
- Liu, R.; Mori, S.; Wake, H.; Zhang, J.; Liu, K.; Izushi, Y.; Takahashi, H.K.; Peng, B.; Nishibori, M. Establishment of in vitro binding assay of high mobility group box-1 and S100A12 to receptor for advanced glycation endproducts: Heparin’s effect on binding. Acta Med. Okayama. 2009, 63, 203–211. [Google Scholar]
- Yang, H.; Wang, H.; Andersson, U. Targeting Inflammation Driven by HMGB1. Front Immunol. 2020, 20, 484. [Google Scholar] [CrossRef]
- Zhu, S.; Ashok, M.; Li, J.; Li, W.; Yang, H.; Wang, P.; Tracey, K.J.; Sama, A.E.; Wang, H. Spermine protects mice against lethal sepsis partly by attenuating surrogate inflammatory markers. Mol. Med. 2009, 15, 275–282. [Google Scholar] [CrossRef]
- Fan, J.; Li, Y.; Levy, R.M.; Fan, J.J.; Hackam, D.J.; Vodovotz, Y.; Yang, H.; Tracey, K.J.; Billiar, T.R.; Wilson, M.A. Hemorrhagic shock induces NAD(P)H oxidase activation in neutrophils: Role of HMGB1-TLR4 signaling. J. Immunol. 2007, 178, 6573–6580. [Google Scholar] [CrossRef]
- Fiuza, C.; Bustin, M.; Talwar, S.; Tropea, M.; Gerstenberger, E.; Shelhamer, J.H.; Suffredini, A.F. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood 2003, 101, 2652–2660. [Google Scholar] [CrossRef]
- Park, J.S.; Svetkauskaite, D.; He, Q.; Kim, J.Y.; Strassheim, D.; Ishizaka, A.; Abraham, E. Involvement of TLR 2 and TLR 4 in cellular activation by high mobility group box 1 protein (HMGB1). J. Biol. Chem. 2004, 279, 7370–7377. [Google Scholar] [CrossRef] [PubMed]
- Kokkola, R.; Andersson, A.; Mullins, G.; Ostberg, T.; Treutiger, C.J.; Arnold, B.; Nawroth, P.; Andersson, U.; Harris, R.A.; Harris, H.E. RAGE is the Major Receptor for the Proinflammatory Activity of HMGB1 in Rodent Macrophages. Scand. J. Immunol. 2005, 61, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Treutiger, C.J.; Mullins, G.E.; Johansson, A.S.; Rouhiainen, A.; Rauvala, H.M.; Erlandsson-Harris, H.; Andersson, U.; Yang, H.; Tracey, K.J.; Andersson, J.; et al. High mobility group 1 B-box mediates activation of human endothelium. J. Intern. Med. 2003, 254, 375–385. [Google Scholar] [CrossRef] [PubMed]
- Lv, B.; Wang, H.; Tang, Y.; Fan, Z.; Xiao, X.; Chen, F. High-mobility group box 1 protein induces tissue factor expression in vascular endothelial cells via activation of NF-kappaB and Egr-1. Thromb. Haemost. 2009, 102, 352–359. [Google Scholar] [PubMed]
- Hori, O.; Brett, J.; Slattery, T.; Cao, R.; Zhang, J.; Chen, J.X.; Nagashima, M.; Lundh, E.R.; Vijay, S.; Nitecki, D. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J. Biol. Chem. 1995, 270, 25752–25761. [Google Scholar] [CrossRef]
- Deng, M.; Tang, Y.; Li, W.; Wang, X.; Zhang, R.; Zhang, X.; Zhao, X.; Liu, J.; Tang, C.; Liu, Z.; et al. The Endotoxin Delivery Protein HMGB1 Mediates Caspase-11-Dependent Lethality in Sepsis. Immunity 2018, 49, 740–753. [Google Scholar] [CrossRef]
- Degryse, B.; Bonaldi, T.; Scaffidi, P.; Muller, S.; Resnati, M.; Sanvito, F.; Arrigoni, G.; Bianchi, M.E. The high mobility group (HMG) boxes of the nuclear protein HMG1 induce chemotaxis and cytoskeleton reorganization in rat smooth muscle cells. J. Cell. Biol. 2001, 152, 1197–1206. [Google Scholar] [CrossRef]
- Yang, D.; Chen, Q.; Yang, H.; Tracey, K.J.; Bustin, M.; Oppenheim, J.J. High mobility group box-1 protein induces the migration and activation of human dendritic cells and acts as an alarmin. J. Leukoc. Biol. 2007, 81, 59–66. [Google Scholar] [CrossRef]
- Dumitriu, I.E.; Bianchi, M.E.; Bacci, M.; Manfredi, A.A.; Rovere-Querini, P. The secretion of HMGB1 is required for the migration of maturing dendritic cells. J. Leukoc. Biol. 2007, 81, 84–91. [Google Scholar] [CrossRef]
- Xu, J.; Jiang, Y.; Wang, J.; Shi, X.; Liu, Q.; Liu, Z.; Li, Y.; Scott, M.J.; Xiao, G.; Li, S.; et al. Macrophage endocytosis of high-mobility group box 1 triggers pyroptosis. Cell. Death. Differ. 2014, 21, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Tadie, J.M.; Bae, H.B.; Jiang, S.; Park, D.W.; Bell, C.P.; Yang, H.; Pittet, J.F.; Tracey, K.; Thannickal, V.J.; Abraham, E.; et al. HMGB1 promotes neutrophil extracellular trap formation through interactions with Toll-like receptor 4. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2013, 304, L342–L349. [Google Scholar] [CrossRef] [PubMed]
- Tadié, J.M.; Bae, H.B.; Banerjee, S.; Zmijewski, J.W.; Abraham, E. Differential activation of RAGE by HMGB1 modulates neutrophil-associated NADPH oxidase activity and bacterial killing. Am. J. Physiol. Cell. Physiol. 2012, 302, C249–C256. [Google Scholar] [CrossRef]
- Gregoire, M.; Tadie, J.M.; Uhel, F.; Gacouin, A.; Piau, C.; Bone, N.; Le, T.Y.; Abraham, E.; Tarte, K.; Zmijewski, J.W. Frontline Science: HMGB1 induces neutrophil dysfunction in experimental sepsis and in patients who survive septic shock. J. Leukoc. Biol. 2017, 101, 1281–1287. [Google Scholar] [CrossRef] [PubMed]
- Robert, S.M.; Sjodin, H.; Fink, M.P.; Aneja, R.K. Preconditioning with high mobility group box 1 (HMGB1) induces lipoteichoic acid (LTA) tolerance. J. Immunother. 2010, 33, 663–671. [Google Scholar] [CrossRef]
- Aneja, R.K.; Tsung, A.; Sjodin, H.; Gefter, J.V.; Delude, R.L.; Billiar, T.R.; Fink, M.P. Preconditioning with high mobility group box 1 (HMGB1) induces lipopolysaccharide (LPS) tolerance. J. Leukoc. Biol. 2008, 84, 1326–1334. [Google Scholar] [CrossRef]
- Patel, V.S.; Sitapara, R.A.; Gore, A.; Phan, B.; Sharma, L.; Sampat, V.; Li, J.; Yang, H.; Chavan, S.S.; Wang, H.; et al. HMGB1 Mediates Hyperoxia-Induced Impairment of Pseudomonas aeruginosa Clearance and Inflammatory Lung Injury in Mice. Am. J. Respir. Cell Mol. Biol. 2012, 48, 280–287. [Google Scholar] [CrossRef]
- Wild, C.A.; Bergmann, C.; Fritz, G.; Schuler, P.; Hoffmann, T.K.; Lotfi, R.; Westendorf, A.; Brandau, S.; Lang, S. HMGB1 conveys immunosuppressive characteristics on regulatory and conventional T cells. Int. Immunol. 2012, 24, 485–494. [Google Scholar] [CrossRef]
- Li, W.; Zhu, S.; Li, J.; D’Amore, J.; D’Angelo, J.; Yang, H.; Wang, P.; Tracey, K.J.; Wang, H. Serum Amyloid A Stimulates PKR Expression and HMGB1 Release Possibly through TLR4/RAGE Receptors. Mol. Med. 2015, 21, 515–525. [Google Scholar] [CrossRef]
- Niemi, K.; Teirila, L.; Lappalainen, J.; Rajamaki, K.; Baumann, M.H.; Oorni, K.; Wolff, H.; Kovanen, P.T.; Matikainen, S.; Eklund, K.K. Serum amyloid A activates the NLRP3 inflammasome via P2X7 receptor and a cathepsin B-sensitive pathway. J. Immunol. 2011, 186, 6119–6128. [Google Scholar] [CrossRef]
- Ather, J.L.; Ckless, K.; Martin, R.; Foley, K.L.; Suratt, B.T.; Boyson, J.E.; Fitzgerald, K.A.; Flavell, R.A.; Eisenbarth, S.C.; Poynter, M.E. Serum amyloid A activates the NLRP3 inflammasome and promotes Th17 allergic asthma in mice. J. Immunol. 2011, 187, 64–73. [Google Scholar] [CrossRef]
- Chen, G.; Li, J.; Qiang, X.; Czura, C.J.; Ochani, M.; Ochani, K.; Ulloa, L.; Yang, H.; Tracey, K.J.; Wang, P.; et al. Suppression of HMGB1 release by stearoyl lysophosphatidylcholine:an additional mechanism for its therapeutic effects in experimental sepsis. J. Lipid. Res. 2005, 46, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Pisetsky, D.S. The Role of IFN-{alpha} and Nitric Oxide in the Release of HMGB1 by RAW 264.7 Cells Stimulated with Polyinosinic-Polycytidylic Acid or Lipopolysaccharide. J. Immunol. 2006, 177, 3337–3343. [Google Scholar] [PubMed]
- Xie, M.; Yu, Y.; Kang, R.; Zhu, S.; Yang, L.; Zeng, L.; Sun, X.; Yang, M.; Billiar, T.R.; Wang, H.; et al. PKM2-dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat. Commun. 2016, 7, 13280. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Xie, M.; Yang, M.; Yu, Y.; Zhu, S.; Hou, W.; Kang, R.; Lotze, M.T.; Billiar, T.R.; Wang, H.; et al. PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nat. Commun. 2014, 5, 4436. [Google Scholar] [CrossRef]
- Zhu, S.; Wang, Y.; Chen, W.; Li, W.; Wang, A.; Wong, S.; Bao, G.; Li, J.; Yang, H.; Tracey, K.J.; et al. High-Density Lipoprotein (HDL) Counter-Regulates Serum Amyloid A (SAA)-Induced sPLA2-IIE and sPLA2-V Expression in Macrophages. PLoS ONE 2016, 11, e0167468. [Google Scholar] [CrossRef]
- Freeman, L.; Guo, H.; David, C.N.; Brickey, W.J.; Jha, S.; Ting, J.P. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J. Exp. Med. 2017, 214, 1351–1370. [Google Scholar] [CrossRef] [PubMed]
- Salina, A.C.G.; Brandt, S.L.; Klopfenstein, N.; Blackman, A.; Bazzano, J.M.R.; Sá-Nunes, A.; Byers-Glosson, N.; Brodskyn, C.; Tavares, N.M.; Da Silva, I.B.S.; et al. Leukotriene B(4) licenses inflammasome activation to enhance skin host defense. Proc. Natl. Acad. Sci. USA 2020, 117, 30619–30627. [Google Scholar] [CrossRef]
- Chaves, M.M.; Sinflorio, D.A.; Thorstenberg, M.L.; Martins, M.D.A.; Moreira-Souza, A.C.A.; Rangel, T.P.; Silva, C.L.M.; Bellio, M.; Canetti, C.; Coutinho-Silva, R. Non-canonical NLRP3 inflammasome activation and IL-1β signaling are necessary to L. amazonensis control mediated by P2X7 receptor and leukotriene B4. PLoS. Pathog. 2019, 15, e1007887. [Google Scholar] [CrossRef]
- Li, W.; Bao, G.; Chen, W.; Qiang, X.; Zhu, S.; Wang, S.; He, M.; Ma, G.; Ochani, M.; Al-Abed, Y.; et al. Connexin 43 Hemichannel as a Novel Mediator of Sterile and Infectious Inflammatory Diseases. Sci. Rep. 2018, 8, 166–18452. [Google Scholar] [CrossRef]
- Dosch, M.; Zindel, J.; Jebbawi, F.; Melin, N.; Sanchez-Taltavull, D.; Stroka, D.; Candinas, D.; Beldi, G. Connexin-43-dependent ATP release mediates macrophage activation during sepsis. Elife 2019, 8, e42670. [Google Scholar] [CrossRef] [PubMed]
- Beyer, E.C.; Steinberg, T.H. Evidence that the gap junction protein connexin-43 is the ATP-induced pore of mouse macrophages. J. Biol. Chem. 1991, 266, 7971–7974. [Google Scholar] [CrossRef]
- Kang, J.; Kang, N.; Lovatt, D.; Torres, A.; Zhao, Z.; Lin, J.; Nedergaard, M. Connexin 43 hemichannels are permeable to ATP. J. Neurosci. 2008, 28, 4702–4711. [Google Scholar] [CrossRef]
- Hulsmans, M.; Clauss, S.; Xiao, L.; Aguirre, A.D.; King, K.R.; Hanley, A.; Hucker, W.J.; Wulfers, E.M.; Seemann, G.; Courties, G.; et al. Macrophages Facilitate Electrical Conduction in the Heart. Cell 2017, 169, 510–522. [Google Scholar] [CrossRef] [PubMed]
- Westphalen, K.; Gusarova, G.A.; Islam, M.N.; Subramanian, M.; Cohen, T.S.; Prince, A.S.; Bhattacharya, J. Sessile alveolar macrophages communicate with alveolar epithelium to modulate immunity. Nature 2014, 506, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Al-Ghadban, S.; Kaissi, S.; Homaidan, F.R.; Naim, H.Y.; El-Sabban, M.E. Cross-talk between intestinal epithelial cells and immune cells in inflammatory bowel disease. Sci. Rep. 2016, 6, 29783. [Google Scholar] [CrossRef]
- Jara, P.I.; Boric, M.P.; Saez, J.C. Leukocytes express connexin 43 after activation with lipopolysaccharide and appear to form gap junctions with endothelial cells after ischemia-reperfusion. Proc. Natl. Acad. Sci. USA 1995, 92, 7011–7015. [Google Scholar] [CrossRef]
- Wong, C.W.; Christen, T.; Kwak, B.R. Connexins in leukocytes: Shuttling messages? Cardiovasc. Res. 2004, 62, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Scheckenbach, K.E.; Crespin, S.; Kwak, B.R.; Chanson, M. Connexin channel-dependent signaling pathways in inflammation. J. Vasc. Res. 2011, 48, 91–103. [Google Scholar] [CrossRef]
- Kang, R.; Zeng, L.; Zhu, S.; Xie, Y.; Liu, J.; Wen, Q.; Cao, L.; Xie, M.; Ran, Q.; Kroemer, G.; et al. Lipid Peroxidation Drives Gasdermin D-Mediated Pyroptosis in Lethal Polymicrobial Sepsis. Cell Host. Microbe. 2018, 24, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zeng, L.; Zhu, S.; Liu, J.; Zeh, H.J.; Kroemer, G.; Wang, H.; Billiar, T.R.; Jiang, J.; Tang, D.; et al. cAMP metabolism controls caspase-11 inflammasome activation and pyroptosis in sepsis. Sci. Adv. 2019, 5, eaav5562. [Google Scholar] [CrossRef]