Neurodegenerative diseases are aging-associated chronic pathological conditions affecting primarily neurons in humans. Inclusion bodies containing misfolded proteins have emerged as a common pathological feature for these diseases. In many cases, misfolded proteins produced by neurons can be transmitted to a neighboring neuron or a non-neuronal cell, leading to the propagation of disease-associated pathology. While undergoing intercellular transmission, misfolded proteins released from donor cells can often change the physiological state of recipient cells. Accumulating evidence suggests that astrocytes are highly sensitive to neuron-originated proteotoxic insults, which convert them into an active inflammatory state (reactive astrogliosis). Conversely, activated astrocytes can release a plethora of factors to impact neuronal functions.
1. Introduction
Neurodegeneration refers to the progressive loss of structure and function of neurons in pathological conditions. Depending on the type and location of the affected neurons, neurodegenerative diseases can display heterogeneous clinical and pathological expressions
[1]. Although research in the past has long been ‘neurocentric’, recent studies have started to shift the paradigm as new roles by glial cells in neurodegenerative diseases are being revealed.
Glial cells were first reported in 1856 by a pathologist named Rudolf Virchow in the book ’Cellular Pathology’. Derived from the ancient Greek word “glía” (meaning “glue” in English), the name “Glia” suggests these cells as “glue” that holds neurons together. However, this view has changed significantly in recent years as more and more neuronal supporting functions have been identified for glial cells.
Glial cells are historically categorized into two main groups: macroglia and microglia. The former includes astrocytes, oligodendrocytes, NG2-glia, and ependymal cells, while microglia are resident phagocytes of the central nervous system (CNS). Among these cell types, astrocytes have drawn significant attention recently due to their unique neuron-safeguarding functions. As the most abundant non-neuronal cells in the CNS, astrocytes are capable of responding to many neurodegeneration-associated events such as metabolic fluctuation, molecular damage, and energy and ion homeostasis disruption
[2]. Additionally, as immune-responding cells, astrocytes also participate in neuroinflammation
[3]. These functions are all tightly regulated during ageing and ageing-associated neurodegeneration.
2. Astrocytes in Tauopathies
2.1. Tau and Tauopathies
Intracellular neurofibrillary tangles (NFTs) formed by hyperphosphorylated Tau are a pathological hallmark of a broad spectrum of neurodegenerative disorders collectively referred to as tauopathies
[4][5][6][4,5,6]. Tauopathies are conventionally classified into two groups. Primary tauopathies, which include progressive supranuclear palsy (PSP), frontotemporal dementia parkinsonism linked to chromosome17 (FTDP-17), Pick’s disease (PiD), corticobasal degeneration (CBD), chronic traumatic encephalopathy (CTE), and argyrophilic grain disease (AGD), refer to disease conditions in which Tau deposit is the predominant pathology
[4][7][4,7]. By contrast, secondary tauopathies involve other pathogenic drivers in addition to Tau deposition. For example, Alzheimer’s disease (AD), the most prevalent cause of dementia, is a secondary tauopathy because it also involves extracellular deposition of amyloid-β (Aβ) plaques
[8][9][8,9].
Tau is a microtubule-binding protein predominantly expressed in neurons in the brain
[10][11][10,11]. However, Tau deposits are prevalent in both neuronal and non-neuronal cells in tauopathies. Immunohistochemistry analyses of phosphorylated Tau revealed six distinct astroglial phenotypes associated with tauopathies including astrocytic plaques (AP), tufted astrocytes (TA), ramified astrocytes (RA), and globular astroglial inclusions (GAI) in primary tauopathies, and thorn shaped astrocytes (TSA) and granular/fuzzy astrocytes (GFA) in aging-related Tau astrogliopathy (ARTAG)
[12][13][14][15][12,13,14,15].
The expression of Tau is regulated by alternative splicing of the Tau-encoding gene MAPT
[16]. The resulting six isoforms contain either 3 or 4 microtubule-binding repeats (referred to as 3R and 4R, respectively) combined with zero to two amino-terminal insertions (NT). Healthy adults express approximately equal amounts of 3R- and 4R-Tau, and aggregates composed of either 3R or 4R Tau have been seen in different tauopathies. However, sporadic tauopathies such as PSP, CBD, FTDP-17, and AGD feature NFT deposits exclusively composed of 4R-Tau
[14].
Post-translational modifications (PTMs) of Tau such as phosphorylation, acetylation, ubiquitination, SUMOylation, methylation, and glycation have long been recognized as a critical contributing factor to tauopathies
[17][18][19][20][21][17,18,19,20,21]. Tau PTMs may enable the formation of the highly ordered β-sheet structures, which facilitates the formation of filamentous Tau inclusions, as indicated by a recent study that reported a role of Tau ubiquitination in filament formation and strain specification
[22]. PTMs may also control Tau stability, and thus influence Tau pathology, as exemplified by the implication of ubiquitin ligases and deubiquitinases (DUB) in Tau stability regulation
[23][24][25][23,24,25]. Among reported PTMs, Tau hyperphosphorylation is thought to be the most significant driving force of tauopathy, possibly because this modification changes the affinity of Tau to microtubule, and thus its aggregation propensity. Noticeably, Tau phosphorylation was also seen in astrocytes, implying a potential role in reactive astrogliosis
[26].
2.2. Astrocytes as a Modulator of AD and Tauopathies
Although most tauopathies including late-onset AD-associated tauopathies arise sporadically within the population, genome-wide association study (GWAS) have identified many tauopathy-associated single-nucleotide polymorphism (SNP) markers
[27][28][29][27,28,29]. Intriguingly, many genes associated with increased risk of neurodegeneration are glial genes (
Table 1).
Table 1. A list of astrocyte- or microglia-specific AD and tauopathy modulators.
| Gene |
Glia Cell Type |
Pathway |
Effect on Aβ |
Effect on Tau |
| APOE [30] |
Astrocyte |
Lipid metabolism, immune response |
Aβ clearance [31][34] |
Tau aggregation and toxicity [32][33][38,39] |
| CLU(APOJ) [34][35][40,41] |
Astrocyte |
Lipid metabolism, immune response |
Amyloid formation [36][42] |
Unknown |
| FERMT2 [37][43] |
Astrocyte |
Integrin signaling, and cell adhesion, angiogenesis |
Aβ production [38][44] |
Tau proteostasis [39][45] |
| WWOX [29] |
Astrocyte |
Putative oxidoreductase, neuronal differentiation [40][46] |
Aβ aggregation [41][47] |
Tau phosphorylation, NFT formation [41][42][47,48] |
| IL1RAP [43][49] |
Astrocyte, oligodendrocyte |
Neuronal synaptogenesis [44][50] |
Unknown |
Unknown |
| PTK2B [45][51] |
Microglia, astrocyte |
Immune response, endocytosis, synaptic transmission |
Unknown |
Tau toxicity [46][52] |
| SORL1 [47][53] |
Microglia, astrocyte |
Endosomal traffic |
APP trafficking [48][54] |
Unknown |
| CELF1 [49][55] |
Astrocyte, oligodendrocyte, microglia |
Unknown |
Unknown |
Unknown |
| EPHA1 [50][56[51],57] |
Astrocyte, oligodendrocyte, microglia |
Cell migration and proliferation, immune response |
Unknown |
Tau toxicity [46][52] |
| CD2AP [50][51][56,57] |
Astrocyte, oligodendrocyte, microglia |
Neurite structure modulation and blood-brain barrier integrity |
Aβ production [52][53][58,59] |
Tau toxicity [54][60] |
ApoE is the strongest genetic risk locus for AD. ApoE E4 carriers have enhanced AD pathology, accelerated cognitive decline and worsened memory performance compared to noncarriers
[30]. As a secreted lipid transport protein that moves lipids between organs, ApoE is expressed primarily in a subset of astrocytes in the CNS
[55][56][31,32]. The mechanism by which ApoE variants alter AD pathology is complex, which is likely linked to the deposition and clearance of Aβ in the brain
[57][31][58][59][60][33,34,35,36,37].
Given the tight link between AD and tauopathy, the role of ApoE in tauopathies has also been examined. By crossing the P301S Tau transgenic mice to those bearing a human ApoE knock-in allele or lacking ApoE completely, Shi et al. showed that P301S/ApoE E4 mice had significantly higher levels of intracellular Tau, more microglia activation and reactive astrogliosis compared to P301S mice bearing other ApoE variants, while the P301S mice lacking ApoE completely had the least tauopathy
[32][38]. More recently, the same group found that astrocyte specific removal of ApoE E4 allele markedly decreased phosphorylated Tau and Tau-associated neurodegeneration, which suggested that astrocyte-derived ApoE4 is a major regulator of tauopathies
[61]. However, another study suggested that neuronal ApoE expression is linked to MHC-I upregulation, which causes tauopathy and selective neurodegeneration
[62].
CLU gene variants (encoding ApoJ/Clusterin) are another strong genetic risk factor for late-onset AD, as established by GWAS
[34][35][40,41]. Like ApoE, CLU is an apolipoprotein predominantly expressed in astrocytes in the brain
[63]. As an extracellular chaperone, CLU secreted by astrocytes can bind to Aβ to prevent Aβ aggregation
[64][65][66][64,65,66]. Accordingly, it has been proposed that increased CLU in glia may be protective in AD and tauopathies
[67].
Other AD risk factors identified by GWAS include FERMT2 (encoding Kindlin-2)
[37][43] and WWOX
[29]. FERMT2 is mainly expressed in astrocytes
[68] but can also be detected in human induced pluripotent stem cell (iPSC)-derived neurons
[39][45]. It is localized to focal adhesions where it interacts with and activates β3 integrin
[69]. The role of FERMT2 in AD and tauopathy is largely unknown, but a genome-wide siRNA screen suggested that FERMT2 may increase Aβ peptide production by elevating the levels of mature APP at the cell surface via membrane recycling
[38][44]. Another candidate-based screening found that knockdown of FERMT2 led to a reduction of phosphorylated Tau
[39][45]. WWOX, encoding a putative oxidoreductase, is expressed in both astrocytes and neurons
[29]. WWOX regulates Aβ aggregation and also binds to Tau to influence Tau hyperphosphorylation and neurofibrillary formation
[41][42][47,48]. Taken together, these genome-wide studies not only identified genetic risk factors for AD and related tauopathies, but also underscored a role for glial cells, especially astrocytes, in driving Aβ- and Tau-associated neuropathology.
2.3. Astrocyte in the Propagation of Tauopathies
An unusual characteristic of tauopathies is the prion-like propagation of Tau-containing fibrils, which correlates with cognition decline and disease progression. Braak and colleagues first reported the spatial and temporal dynamics of Tau-containing fibrils in AD brains. Specifically, NFTs, first uncovered in the transentorhinal region, appear to traverse along several anatomical paths to reach the hippocampus and eventually the neocortex region
[70]. The progressive spreading of Tau inclusions was later recapitulated in mouse models
[71][72][73][74][71,72,73,74]. There is now comprehensive evidence that supports the idea that pathogenic Tau species undergo cell-to-cell transmission with a prion-like property
[75][76][77][78][75,76,77,78]. However, the ultimate spatial distribution of Tau NFTs is distinct among tauopathies due to strain distinctions. Additionally, external factors may also influence the spreading pattern of tauopathy. For example, in AD, genetic and clinical evidence indicates that Aβ plaque deposition can facilitate the spreading of tauopathy
[79][80][81][79,80,81]. Moreover, Tau-containing aggregates accumulated in glial cells (both microglia and astrocytes) may also modulate Tau transmission (see below).
The intercellular transmission of Tau is likely initiated when neurons release Tau either in monomeric or small oligomerized forms. Indeed, Tau is readily detected in the interstitial fluid (ISF) of the brain under normal conditions
[82]. Accumulating evidence suggests that Tau species can be released from neurons independent of cell death, and this process is modulated by neuronal activities
[83][84][85][83,84,85]. The mechanisms underlying Tau release are controversial. Specifically, some studies showed that Tau is predominantly released in a free soluble form
[86][87][88][89][86,87,88,89] but other studies suggested membrane-associated vesicles such as exosome as an extracellular Tau carrier
[90][91][90,91]. It is possible that multiple mechanisms coexist to regulate Tau secretion.
Once in the cell exterior, Tau may be taken up by cells via endocytosis
[92], micropinocytosis
[93] or other forms of cargo internalization
[94]. One study suggests that healthy neurons efficiently take up both normal and aggregated Tau by distinct but overlapping mechanisms, which indicates the existence of multiple Tau receptors for internalization
[95]. Not only neurons, but other cell types in the brain such as microglia and astrocytes can also engulf Tau proteins
[33][93][96][39,93,96]. In certain immortalized cells, endocytosis of Tau preformed fibrils (PFFs) is initiated when Tau binds to the cell surface heparan sulfate proteoglycans (HSPGs)
[94][97][98][94,97,98], which cooperate with a membrane receptor to mediate Tau internalization
[99]. However, HSPG does not play a major role in Tau uptake in primary astrocytes
[99][100][99,100]. We recently used a spatially resolved proteomic mapping strategy to identify the integrin αV/β1 complex as a receptor that binds human Tau fibrils to mediate their entry into astrocytes
[33][39]. When inside the astrocyte, Tau may be cleared by lysosomal degradation or the recently reported astrocytic glymphatic system
[101].
Although Tau aggregates have been observed in various cell types in the brain, most attention in the field has been given to intraneuronal or extracellular Tau deposits, while the glial involvement was rarely considered. This deficiency may significantly hinder our understanding of the mechanisms underlying the transmission of tauopathy. To better understand the role of glial Tau deposits in tauopathy, the following questions need to be carefully addressed. (i) Which glial cell type accumulates the most pathological Tau in tauopathies? (ii) Which Tau species is propagated in each tauopathy and how is their distribution in the brain sculpted? (iii) Do astrocytes or other glial cells contribute to Tau propagation? (iv) Does the accumulation of Tau in astrocytes contribute to neurodegeneration, and if so, what is the underlying mechanism?
To date, only a few published studies attempted to address these questions, which collectively paint an incomplete model. Tau accumulation in astrocytes was reported in some tauopathy mouse models
[102][103][102,103]. More recently, using an in vivo reporter system, Anastasie et al. demonstrated bidirectional exchanges of Tau protein between neuron and astrocyte. They further showed that soluble Tau, but not Tau aggregates, is toxic to a subpopulation of hippocampal astrocytes
[104]. This study hints at a role for astrocytes in tauopathy. A few studies investigated the disease relevance of astrocytic Tau in other experimental models. For example, expression of human Tau in glia in a Drosophila model led to neurotoxicity, suggesting that Tau, if propagated into glial cells, might have a pathogenic activity
[105]. Likewise, in a transgenic mouse model, astrocyte-specific expression of human Tau leads to neurodegeneration
[106]. A study by Richetin et al. also suggests astrocytic Tau as a causal factor for dementia. They detected Tau accumulation in astrocytes of the hilus, a portion of the hippocampus in AD patients; in mice, overexpression of the 3R Tau variant in hilar astrocytes of the dentate gyrus impaired mitochondrial function and thus ATP production
[107]. Intriguingly, this work detected 3R Tau in astrocytes, unlike previous studies that attributed astrocytic Tau deposits predominantly to the 4R isoform
[108].
Two recent papers further link Tau to the build-up of astrocytic senescent cells in the brain, which contribute to neurodegeneration. Musi et al. showed that destroying senescent cells in mice at early stages of tauopathy slows neurodegeneration and corrects aberrant brain blood flow
[109], whereas Bussian et al. reported that specific elimination of senescent astrocytes is sufficient to prevent neurodegeneration and cognitive decline in a mouse model of tauopathy
[110]. Although these studies both hinted at a critical role for astrocytic Tau in cell senescence, which in turn influences neurodegeneration, how the senescent state of microglia or astrocytes is aligned with other tauopathy-related features remains unclear. Altogether, the existing evidence suggests that in tauopathies, Tau proteopathy may exist beyond neurons, which warrants additional studies.
2.4. Tauopathies Are Associated with Widespread Reactive Astrogliosis
Under neurodegeneration conditions, astrocytes also undergo significant changes, which can fall into three morphologically defined categories: (i) atrophy/degeneration occurs as astrocytes lose their homeostatic function to support neuronal growth. (ii) Astroglial remodeling refers to morphologic alterations of astrocytes under disease or CNS injury conditions. (iii) Reactive astrogliosis refers to special responses of astrocytes to different insults in many CNS disorders, which result in astroglial hypertrophy (increased volume, thickened processes, and increased expression of GFAP etc.
[111][112][111,112]).
Due to their sensitivity to the brain environment, astrocytes can enter a “reactive” or “activated” state now generally termed astrogliosis
[113]. Many markers of reactive astrocytes
[2][114][115][2,114,115] have been identified and used to characterize the neurodegenerative disease states. Under certain experimental conditions, reactive astrogliosis induced by lipopolysaccharide (LPS) increases the phagocytic activity of astrocytes, which may mitigate tauopathies if the activated astrocytes help to clear protein aggregates
[116][117][116,117]. However, reactive astrogliosis under pathophysiological conditions can also be a major contributor of chronic neuroinflammation (
Figure 1), which exacerbates neurodegeneration in several animal disease models
[118][119][118,119]. Thus, it seems that upon activation, astrocytes might be transformed into multiple functional states, resulting in a heterogeneous population.
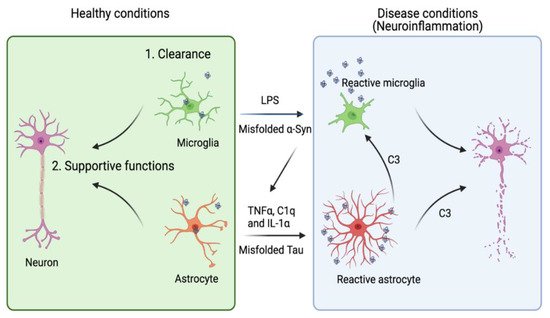
Figure 1. A dual role of microglia and astrocytes in neuronal growth and neurodegenerative diseases. Accumulating evidence suggests that the activation of microglia and astrocytes may be a double-edged sword. Under healthy conditions, microglia and astrocytes engulf neuron-derived misfolded proteins such as Tau and α-syn to promote protein homeostasis in the brain micro-environment. Astrocytes can also provide other supportive functions including axonal guidance and synaptic support. However, when these cells are overactivated by toxic factors (e.g., LPS or excess amount of extracellular Tau or α-syn), they release pro-inflammatory cytokines and chemokines to disrupt neuronal integrity. Reactive microglia can also cross-activate astrocytes by releasing cytokines such as TNFα, IL1-α and C1q. Conversely, astrocytes release complement C3, which can act on both microglia and neurons to further enhance neuroinflammation. Image created in BioRender.com.
A recent transcription profiling study identified two gene expression signatures corresponding to two functional states of reactive astrogliosis termed as A1 and A2, respectively. A2 astrocytes have a neuron-supporting function and can restore neuronal activities after injury. By contrast, A1 astrocytes not only fail to promote synapse formation, but also release some neurotoxic factors. Complement C3 was later identified as an astrocyte-released factor that induces neuronal impairment, possibly through a C3 receptor (C3aR) because C3aR1 deficiency reverses plaque-proximal synapse loss in a Tau P301S mouse model
[120]. Interestingly, astrocyte-released C3 appears to crosstalk to microglia as well, indicating a possible vicious cycle among neuron, astrocyte, and microglia in tauopathy
[121].
The A2 to A1 switch of astrocytes, instigated by microglia, appears to convert astrocytes from a supporter of neuronal homeostasis to a cell death promoter in AD. Interleukin-1α (IL-1α), tumor necrosis factor α (TNFα), and complement component 1q (C1q) secreted from activated microglia were shown to collectively induce the A1 switch phenotype
[122]. Hence, blocking the transformation of astrocytes to the A1 state by these factors may be a potential therapeutic strategy, as suggested by a recent study using a Parkinson’s disease (PD) mouse model
[123]. However, this oversimplified model has recently been challenged. Concerns were raised regarding the potential overlook of astrocytic heterogeneity and the complexity of the factors implicated in shaping the astrocyte phenotypes during disease progression
[124][125][124,125]. Recent advancements in single cell transcriptomics may help better define the various astrocytic states associated with different pathological conditions
[126][127][126,127].
One of the common insults that change astrocyte state in neurodegenerative diseases is abnormal protein aggregates such as Aβ-, Tau-, and α-syn-containing fibrils. For instance, Aβ peptides, derived from abnormal processing of amyloid precursor protein (APP), can form distinct aggregated states, which activate different astrocytic receptors to induce a pro-inflammatory NFκB pathway
[128][129][128,129]. Distinct Tau species also differentially activate integrin signaling in primary mouse astrocytes, which leads to NFκB activation and the release of pro-inflammatory cytokines and chemokines
[33][39]. In rodent models of AD and Huntington’s disease (HD), NFκB activation in astrocytes was observed
[119][130][119,130]. Thus, the NFκB pathway appears to be a critical link that connects extracellular proteotoxic insults to astrogliosis and neuroinflammation.
Besides the NFκB pathway, the Janus kinase/signal transducer and activator of transcription 3 (JAK/STAT3) pathway is also ubiquitously involved in cell proliferation, survival, and differentiation. STAT3 was recently suggested as a mediator of reactive astrogliosis under pathological conditions such as AD and HDs
[131]. However, the contributions of STAT3-mediated reactive astrogliosis to these diseases are not entirely clear. For example, one study suggested that JAK/STAT3 activation is associated with a scar-forming astrocyte activity in a model of acute spinal cord injury
[132], which limits inflammation spreading
[133]. By contrast, in an APP/PS1 model of AD, STAT3 deficient animals showed reduced β-amyloid levels and plaque burden, decreased pro-inflammatory cytokines, and rescued memory decline. Similarly, in a Tau mouse AD model, inhibition of STAT3 also rescues Tau pathology, ameliorates neuroinflammation, and reverses synaptic deficits
[120]. Thus, whether reactive astrogliosis is detrimental or beneficial for damaged neurons may depend on the cause of neurodegeneration.