Blood-brain barrier (BBB) disruption following ischemic stroke (IS) contributes to hemorrhagic transformation, brain edema, increased neural dysfunction, secondary injury, and mortality. Brain endothelial cells form a para and transcellular barrier to most blood-borne solutes via tight junctions (TJs) and rare transcytotic vesicles. The prevailing view attributes the destruction of TJs to the resulting BBB damage following IS, recent studies define a stepwise impairment of the transcellular barrier followed by the paracellular barrier which accounts for the BBB leakage in IS. The increased endothelial transcytosis that has been proven to be caveolae-mediated, precedes and is independent of TJs disintegration. Thus, our understanding of post-stroke BBB deficits needs to be revised, these recent findings could provide a conceptual basis for the development of alternative treatment strategies. Presently, our concept of how BBB endothelial transcytosis develops is incomplete and treatment options remain limited. This entreviewy summarizes the cellular structure and biological classification of endothelial transcytosis at the BBB and reviews the related molecular mechanisms. Meanwhile, relevant transcytosis-targeted therapeutic strategies for IS and research entry points are prospected.
- transcytosis
- caveolae
- blood-brain barrier
- ischemic stroke
1. Introduction
Endothelial transcytosis in the CNS is practically negligible under basal conditions, but a prominent feature following acute neurologic injury. Specifically, the rapid increase of this process in IS brings forth the exact role and contribution of endothelial transcytosis in the formation of a sound functional barrier. Delineating this fundamental question in normal development and physiology provides fundamental understanding and key mechanisms dysregulated in stroke. To this end, Chenghua Gu’s team employed a zebrafish model to investigate the involvement of endothelial transcytosis in a vertebrate BBB development. In leveraging this powerful optically transparent model organism with live EM, Gu et al. visualized in real-time the dynamic formation of the zebrafish BBB [14][1]. Early development of the BBB corresponded with high levels of neural endothelial, a process that is subsequently suppressed later in life. Notably, the timing of suppression of brain endothelial transcytosis coincides with the establishment of BBB function [14][1]. Similarly, in mice, the same group reported that the formation of the functional blood-retinal barrier (BRB) was similarly conditional on the gradual suppression of brain endothelial transcytosis [15][2]. Furthermore, because functional TJs were already established on CNS vasculature, the vascular leakage of the immature BRB was attributed exclusively to the process of endothelial transcytosis [15][2]. Correspondingly, Mfsd2a +/+ and Mfsd2a −/− mutant mice, with either increased or decreased transcytosis, mirrored the aforementioned relationship between BRB integrity and transcytosis levels; elevated transcytosis resulted in delayed BRB sealing and mice with suppressed transcytotic activity displayed earlier BRB sealing [15][2]. Therefore, CNS endothelial transcytosis governs the development of a functional BBB and BRB. The contingent suppression of this process is informed by developmental stage, and time is a principal contributor to the formation of a functional barrier.
2. Cellular Basis of BBB Structure and Endothelial Transcytosis
The remainder of this review briefly summarizes the cellular composition, biological classification, and molecular regulatory mechanisms which underlie BBB endothelial transcytosis. Meanwhile, relevant transcytosis-targeted therapeutic strategies for IS and research entry points are prospected.
The “sandwich” structure of the BBB determines that its constituent components, the ECs, pericytes, and astrocytes, are structurally adjacent and functionally coordinated to control the influx and efflux of substances into the CNS. Pericytes envelop ECs and are embedded within the basement membrane to facilitate extensive and reciprocal intrasignaling. Several influential studies have shown that the precise ratio of pericytes to ECs is critical to BBB integrity; pericytogenesis [16,17,18,19][3][4][5][6] results in BBB impairment, due to increased endothelial transcytosis. In pathological conditions such as IS, an increase of CNS endothelial vesicles is accompanied by a change of coverage from pericytes at the basement membrane, the swelling of astrocytic end-feet and their mitochondrial dysfunction [12,13][7][8] ( Figure 1 ). In young mice, ischemia led to a significant increase in the pericytes process area and vessel coverage over 72 h post ischemia whereas these changes were abrogated with aging. Besides, Caveolae-like vesicles similar to those found in the ECs were also observed in pericytes at 3 and 72 h after ischemia [12][7]. An influential study found that the physiological blood-brain plasma protein uptake is impaired with age by a shift in transcytosis from ligand-specific receptor-mediated to nonspecific caveolar transcytosis, and this age-related shift in transport occurred alongside a specific loss of pericytes coverage [20][9]. These findings indicate that pericytes also produce barrier function by regulating endothelial transcytosis; however, the specific genes in pericytes and the paracrine signal mechanisms regulating barrier function between pericytes and ECs remain undefined.
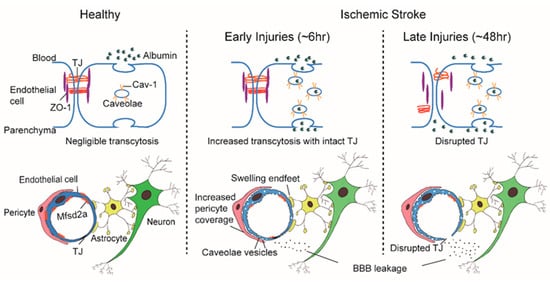
3. Biological Classification of BBB Endothelial Transcytosis
Transcytosis is the vesicular trafficking of molecules between the luminal and abluminal cell membranes. Macromolecules are first endocytosed or internalized by vesicles on one side of the cell, trafficked in vesicles, and then exocytosed or released on the other side of the cell. It remains poorly understood what type of vesicles and different transcytosis mechanisms are involved. How vesicle formation and transcytosis are maintained at low levels in brain ECs is also unknown. The current consensus postulates that transcytosis in CNS endothelial cells can be divided into two categories: receptor-mediated transcytosis (RMT), in which ligand-receptor binding mediates endocytosis such as transferrin and insulin [21[10][11],22], and nonselective adsorptive transcytosis, in which charged interactions between the molecule and plasma membrane facilitates cargo entry, such as with albumin. Interestingly, a recent study with C57BL/6 mouse revealed decreased plasma protein transport activity through the BBB in the aged brain driven by an age-related shift in transport from ligand-specific receptor-mediated to nonspecific caveolar transcytosis [20][9].
The two known major endocytic pathways at the BBB are clathrin-mediated and caveolae-mediated [23][12]. Clathrin-mediated transcytosis is the endocytosis of cargo through clathrin-coated vesicles and is ubiquitous in all cell types [24][13]. Most RMT is clathrin-mediated, such as transferrin and insulin transport [25][14]. A review by Xu et al. [25][14] provides an in-depth description of this complicated process. Prior reports point to the caveolae-mediated mechanism as predominant in CNS endothelial transcytosis after acute IS [11,12,13][15][7][8]. Consequently, IS-induced transcytosis has focused intensely upon alterations of caveolae and relevant proteins. Caveolae are omega-shaped invaginations of the plasma membrane approximately 60–80 nm in length, and are rich in cholesterol and glycosphingolipids [26][16]. The caveolae were first observed by George Palade et al., who also put forth the notion of caveolae vesicles as mass-carriers of fluid and solutes across the ECs [25][14]. Subsequent studies have confirmed Palade’s hypothesis, identifying macromolecular substances like albumin, which are transported via caveolae-mediated transcytosis [27][17]. Conversely, caveolae-deficient ECs showed deficiencies in the uptake and transport of albumin in periphery [28][18] and CNS [20][9].
Caveolin and cavin play a key role in the formation of caveolae. Cavin is an adaptive protein that forms oligomers crucial for membrane bending, and caveolin is an integral membrane protein that binds to cytosolic cavin to form small vesicles [29,30][19][20]. Caveolin (Cav) can be subdivided into three groups based on their interactive functions with cavin: Cav-1, Cav-2, and Cav-3. The latest data show that Cav-1 and cavin-1 act synergistically to generate a unique lipid environment in caveolae [31][21]. In the brain, Cav-1 and Cav-2 are mainly expressed in ECs, Cav-3 is expressed in astrocytes, and notably, only Cav-1 is considered to be essential for caveolae formation [32,33][22][23].
4. Molecular Regulatory Mechanisms of Caveolae-Mediated Transcytosis in IS
Increasingly, studies associate Cav-1 with the permeabilized BBB following various pathological conditions, including IS. It has been found that Cav-1 expression increases in the rat cortical at 12 h after hypothermic brain injury, preceding the decreased expression of occludin and claudin-5 at 48 h [34][24]. Moreover, Phoneutria nigriventer spider venom (PNV)-induced BBB breakdown and increased vesicle trafficking was related to increased Cav-1α expressions in cerebellar capillaries and Purkinje neurons at 2, 5, and 24 h after brain injury. The major fraction of Cav-1 upregulation was localized to the white matter and granular layers [35][25]. The role of Cav-1 is highlighted in cortical spreading depolarizations (CSDs), where injury-induced BBB permeability is specifically mediated by increased endothelial caveolae-mediated transcytosis starting at 3~6 h and lasting up to 24 h [36][26]. Notably, CSD-induced BBB leakage was conspicuously absent in Cav-1 −/− mice compared with age-matched wild-type (WT) controls [36][26]. Knowland et al. report that transcellular, but not paracellular, permeability is reduced in the cortical vessels of Cav-1 null mice at 6 and 27 h after t-MCAO. Cav-1 deficiency only significantly reduced the amount of circulating albumin transported into the brain parenchyma with endosome vesicles routed, but no effects on biocytin-TMR or IgG transportation were detected [11][15]. Moreover, TJ ultrastructural morphology expression levels and subcellular localization of major TJs, claudin-5, occludin, and ZO-1, in healthy brain ECs are indistinguishable between those of Cav-1 −/− mice [11][15].
These findings endorse the notion that upregulation of Cav-1 is functionally related to an increase in BBB permeability via increased endothelial transcytosis. Given the important role of caveolae and Cav-1 in early BBB breakdown following IS, they are attractive targets of modulation to attenuate BBB dysfunction, and thus theoretically provide the downstream benefits of arresting edema formation and infiltration of peripheral immune cells, crucial elements for improving stroke outcomes. Still, it is worth mentioning that studies have also reported that Cav-1 deficiency can aggravate the injury of IS. Work by Knowland et al. supports the divergent detrimental effects of deficiency, showing increased lesion volumes in Cav1 −/− mice compared to WT controls [11][15]. Hirt and colleagues demonstrated a protective role of endogenous Cav-1 in the first week of IS which acted to promote neovascularization, astrogliosis and scar formation [37,38][27][28]. Furthermore, increased expression of Cav-1 was found in new blood vessels in the lesion and peri-infarct areas. Cav1 −/− mice also displayed enhanced hemispheric swelling, lesion volume, and worsened neurological outcomes compared to WT controls. These worsened outcomes coincided with reduced neovascularization and modified astrogliosis, without the formation of proper glial scarring around the lesion at 3 days post injury [37][27], decreased perivascular AQP4 expression in peri-infarct and contralateral cortical regions, and impaired perivascular AQP4 covering [38][28], of stroked Cav1 −/− mice. Gu et al. recently showed that, unlike other vascular segments of ECs in the CNS, arteriolar ECs have abundant caveolae and actively relay signals from the CNS to smooth muscle cells through a caveolae-dependent pathway to realize vasodilation, which is independent of the endothelial NO synthase (eNOS)-mediated NO pathway [39][29]. In patients, a clinical study suggested that the loss of Cav-1 may be associated with the hemorrhagic transformation following IS. Castellanos M. et al. tested the serum Cav-1 levels of 133 first-time stroke patients that underwent thrombolytic therapy with recombinant tissue plasminogen activator (rt-PA) within 4.5 h after the onset of symptoms were detected. Results showed circulating Cav-1 levels in stroke patients were higher than those of healthy controls, while the basic level of serum Cav-1 in patients with substantive and symptomatic hemorrhage was lower than that in other patients. The level of serum Cav-1 in patients with hemorrhagic transformation remained stable within 72 h after stroke, while the level of serum Cav-1 in other patients decreased during this period, suggesting that a low serum Cav-1 level can be used as an independent predictor of hemorrhagic transformation after r-tPA treatment [40][30].
Significant differences differentiate barrier characteristics and vesicle numbers between central and peripheral ECs; whether BBB ECs express a specific set of genes that govern the formation and maintenance is a major outstanding question. Again, innovative work from the Gu team illuminated answers to this question. Gu et al. identified over 200 highly expressed differential genes in cortical ECs that are barely expressed in lung ECs [42][31]. Among these differentially expressed genes, the major facilitator superfamily domain containing 2a (Mfsd2a) stands out as selectively expressed in BBB-containing blood vessels and regulated by pericytes [42][31]. Previously, Mfsd2a was reported to be a transmembrane protein expressed in the placenta and testis, which have highly restrictive barrier properties [43][32]. Mfsd2a mutants display continuous BBB leakage from embryo to adulthood [42][31], and EM imaging shows a dramatic increase in CNS ECs transcytosis without obvious TJs abnormality [14,42][1][31]. Further studies showed that the lipid transport function of Mfsd2a may lead to a significant difference in lipid signaling between the CNS and peripheral ECs; lipids transported by Mfsd2a establishes a unique lipid environment that inhibits caveolae vesicle formation in CNS ECs to suppress transcytosis and ensure BBB integrity [44,45][33][34]. These findings identify Mfsd2a as a key regulator of BBB function that may act by suppressing caveolae-mediated transcytosis in CNS ECs. Based on the reviewed literature, it is our belief that the preclinical experimental data indicate that caveolae-mediated transcytosis is actively inhibited in CNS ECs to ensure BBB integrity, which presents a promising therapeutic target that warrants future study ( Figure 1 ).
Increased BBB permeability is correlated to hemorrhagic transformation, which represents the main limitation of rt-PA thrombolytic therapy in IS. Presently, tPA remains the only approved therapeutic treatment available for IS patients; however, strict inclusion criteria and a limited therapeutic time window limit its ubiquitous usage for patients. Creative strategies to overcome these realities would substantially alter the treatment approach for IS and benefit many patients yearly. Suzuki et al. showed that the transient increase of BBB permeability is related to the endothelial transcytosis in the ischemic area, which is partially regulated by vascular endothelial growth factor (VEGF) [46,47][35][36]. They found that rt-PA treatment at 4 h after MCAO transiently increased BBB permeability, accompanied by increased endothelial transcytosis in ischemic regions that could be inhibited by low-density lipoprotein receptor family (LDLRs) or VEGF receptor-2 (VEGFR-2) antagonists. In the immortalized bEnd.3 ECs ischemic model, rt-PA upregulated VEGF expression and VEGFR-2 phosphorylation in an LDLR-dependent manner. rt-PA treatment further increased monolayer endocytosis and transcytosis of bEnd.3 ECs, which could also be inhibited by antagonists such as LDLR, VEGF, or VEGFR-2. The authors suggested that rt-PA increases the permeability of BBB in IS by inducing VEGF, which partially mediates the subsequent increase of endothelial transcytosis. Although the investigation is still in an early phase, modulating endothelial transcytosis may improve tPA efficacy or tolerability, and studies inhibiting the production of VEGF may complement thrombolytic therapy of rt-PA after stroke [46][35]; Interestingly, recent studies have indicated that besides the most well-studied angiogenic growth factors, the guidance molecules also play a role in post stroke BBB leakage [48][37]. Rust et al. demonstrated that anti-Nogo-A antibodies partially reverse the VEGF-induced BBB leakage when coadministrated through implanted mini-osmotic pumps for seven consecutive days after stroke. Moreover, anti-Nogo-A antibodies have similar proangiogenic effects as local VEGF treatment and do not increase vascular permeability in the peri-infarction regions [49][38]. Whether their mechanism of action is related to the inhibition of endothelial transcytosis remains unclear. However, this does provide a novel therapeutic strategy, and considering the broad impact of expanding tPA access all promising avenues should be investigated.
References
- O’Brown, N.M.; Megason, S.G.; Gu, C. Suppression of transcytosis regulates zebrafish blood-brain barrier function. Elife 2019, 8, e47326.
- Chow, B.W.; Gu, C. Gradual Suppression of Transcytosis Governs Functional Blood-Retinal Barrier Formation. Neuron 2017, 93, 1325–1333.e3.
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes are required for blood–brain barrier integrity during embryogenesis. Nature 2010, 468, 562–566.
- Bell, R.D.; Winkler, E.A.; Sagare, A.P.; Singh, I.; LaRue, B.; Deane, R.; Zlokovic, B.V. Pericytes Control Key Neurovascular Functions and Neuronal Phenotype in the Adult Brain and during Brain Aging. Neuron 2010, 68, 409–427.
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood–brain barrier. Nature 2010, 468, 557–561.
- Reyahi, A.; Nik, A.M.; Ghiami, M.; Gritli-Linde, A.; Pontén, F.; Johansson, B.R.; Carlsson, P. Foxf2 Is Required for Brain Pericyte Differentiation and Development and Maintenance of the Blood-Brain Barrier. Dev. Cell 2015, 34, 19–32.
- Nahirney, P.C.; Reeson, P.; Brown, C.E. Ultrastructural analysis of blood–brain barrier breakdown in the peri-infarct zone in young adult and aged mice. J. Cereb. Blood Flow Metab. 2016, 36, 413–425.
- Haley, M.J.; Lawrence, C.B. The blood–brain barrier after stroke: Structural studies and the role of transcytotic vesicles. J. Cereb. Blood Flow Metab. 2016, 37, 456–470.
- Yang, A.C.; Stevens, M.Y.; Chen, M.B.; Lee, D.P.; Stähli, D.; Gate, D.; Contrepois, K.; Chen, W.; Iram, T.; Zhang, L.; et al. Physiological blood–brain transport is impaired with age by a shift in transcytosis. Nat. Cell Biol. 2020, 583, 425–430.
- Roberts, R.L.; Fine, R.E.; Sandra, A. Receptor-mediated endocytosis of transferrin at the blood-brain barrier. J. Cell Sci. 1993, 104, 521–532.
- Duffy, K.R.; Pardridge, W. Blood-brain barrier transcytosis of insulin in developing rabbits. Brain Res. 1987, 420, 32–38.
- Ayloo, S.; Gu, C. Transcytosis at the blood–brain barrier. Curr. Opin. Neurobiol. 2019, 57, 32–38.
- Pearse, B.M. Clathrin: A unique protein associated with intracellular transfer of membrane by coated vesicles. Proc. Natl. Acad. Sci. USA 1976, 73, 1255–1259.
- Xu, Y.; Xia, J.; Liu, S.; Stein, S.; Ramon, C.; Xi, H.; Wang, L.; Xiong, X.; Zhang, L.; He, D.; et al. Endocytosis and membrane receptor internalization: Implication of F-BAR protein Carom. Front. Biosci. 2017, 22, 1439–1457.
- Knowland, D.; Arac, A.; Sekiguchi, K.J.; Hsu, M.; Lutz, S.; Perrino, J.; Steinberg, G.K.; Barres, B.A.; Nimmerjahn, A.; Agalliu, D. Stepwise Recruitment of Transcellular and Paracellular Pathways Underlies Blood-Brain Barrier Breakdown in Stroke. Neuron 2014, 82, 603–617.
- Echarri, A.; Del Pozo, M.A. Caveolae. Curr. Biol. 2012, 22, R114–R116.
- Preston, J.; Abbott, N.J.; Begley, D.J. Transcytosis of Macromolecules at the Blood–Brain Barrier. In Advances in Pharmacology; Academic Press: Cambridge, MA, USA, 2014; Volume 71, pp. 147–163.
- Schubert, W.; Frank, P.; Razani, B.; Park, D.S.; Chow, C.-W.; Lisanti, M.P. Caveolae-deficient Endothelial Cells Show Defects in the Uptake and Transport of Albumin in Vivo. J. Biol. Chem. 2001, 276, 48619–48622.
- Rothberg, K.G.; Heuser, J.E.; Donzell, W.C.; Ying, Y.-S.; Glenney, J.R.; Anderson, R.G. Caveolin, a protein component of caveolae membrane coats. Cell 1992, 68, 673–682.
- Hill, M.M.; Bastiani, M.; Luetterforst, R.; Kirkham, M.; Kirkham, A.; Nixon, S.; Walser, P.; Abankwa, D.; Oorschot, V.M.; Martin, S.; et al. PTRF-Cavin, a Conserved Cytoplasmic Protein Required for Caveola Formation and Function. Cell 2008, 132, 113–124.
- Zhou, Y.; Ariotti, N.; Rae, J.; Liang, H.; Tillu, V.; Tee, S.; Bastiani, M.; Bademosi, A.T.; Collins, B.M.; Meunier, F.A.; et al. Caveolin-1 and cavin1 act synergistically to generate a unique lipid environment in caveolae. J. Cell Biol. 2021, 220, e202005138.
- Ikezu, T.; Ueda, H.; Trapp, B.D.; Nishiyama, K.; Sha, J.F.; Volonte, D.; Galbiati, F.; Byrd, A.L.; Bassell, G.; Serizawa, H.; et al. Affinity-purification and characterization of caveolins from the brain: Differential expression of caveolin-1, -2, and -3 in brain endothelial and astroglial cell types. Brain Res. 1998, 804, 177–192.
- Virgintino, D.; Robertson, D.; Errede, M.; Benagiano, V.; Tauer, U.; Roncali, L.; Bertossi, M. Expression of caveolin-1 in human brain microvessels. Neuroscience 2002, 115, 145–152.
- Nag, S.; Venugopalan, R.; Stewart, D.J. Increased caveolin-1 expression precedes decreased expression of occludin and claudin-5 during blood–brain barrier breakdown. Acta Neuropathol. 2007, 114, 459–469.
- Soares, E.; Mendonça, M.; Irazusta, S.P.; Coope, A.; Stávale, L.M.; Da Cruz-Höfling, M.A. Evidences of endocytosis via caveolae following blood–brain barrier breakdown by Phoneutria nigriventer spider venom. Toxicol. Lett. 2014, 229, 415–422.
- Sadeghian, H.; Lacoste, B.; Qin, T.; Toussay, X.; Rosa, R.; Oka, F.; Chung, D.; Takizawa, T.; Gu, C.; Ayata, C. Spreading depolarizations trigger caveolin-1-dependent endothelial transcytosis. Ann. Neurol. 2018, 84, 409–423.
- Blochet, C.; Buscemi, L.; Clément, T.; Gehri, S.; Badaut, J.; Hirt, L. Involvement of caveolin-1 in neurovascular unit remodeling after stroke: Effects on neovascularization and astrogliosis. J. Cereb. Blood Flow Metab. 2018, 40, 163–176.
- Filchenko, I.; Blochet, C.; Buscemi, L.; Price, M.; Badaut, J.; Hirt, L. Caveolin-1 Regulates Perivascular Aquaporin-4 Expression After Cerebral Ischemia. Front. Cell Dev. Biol. 2020, 8, 371.
- Chow, B.W.; Nuñez, V.; Kaplan, L.; Granger, A.J.; Bistrong, K.; Zucker, H.L.; Kumar, P.; Sabatini, B.L.; Gu, C. Caveolae in CNS arterioles mediate neurovascular coupling. Nature 2020, 579, 106–110.
- Castellanos, M.; van Eendenburg, C.; Gubern, C.; Kadar, E.; Huguet, G.; Puig, J.; Sobrino, T.; Blasco, G.; Serena, J.; Sánchez, J.M. Low Levels of Caveolin-1 Predict Symptomatic Bleeding After Thrombolytic Therapy in Patients with Acute Ischemic Stroke. Stroke 2018, 49, 1525–1527.
- Ben-Zvi, A.; Lacoste, B.; Kur, E.; Andreone, B.J.; Mayshar, Y.; Yan, H.; Gu, C. Mfsd2a is critical for the formation and function of the blood–brain barrier. Nature 2014, 509, 507–511.
- Esnault, C.; Priet, S.; Ribet, D.; Vernochet, C.; Bruls, T.; Lavialle, C.; Weissenbach, J.; Heidmann, T. A placenta-specific receptor for the fusogenic, endogenous retrovirus-derived, human syncytin-2. Proc. Natl. Acad. Sci. USA 2008, 105, 17532–17537.
- Andreone, B.J.; Chow, B.W.; Tata, A.; Lacoste, B.; Ben-Zvi, A.; Bullock, K.; Deik, A.A.; Ginty, D.D.; Clish, C.; Gu, C. Blood-Brain Barrier Permeability Is Regulated by Lipid Transport-Dependent Suppression of Caveolae-Mediated Transcytosis. Neuron 2017, 94, 581–594.e5.
- Nguyen, L.N.; Ma, D.; Shui, G.; Wong, P.; Cazenave-Gassiot, A.; Zhang, X.; Wenk, M.R.; Goh, E.; Silver, D. Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nature 2014, 509, 503–506.
- Suzuki, Y.; Nagai, N.; Yamakawa, K.; Muranaka, Y.; Hokamura, K.; Umemura, K. Recombinant Tissue-Type Plasminogen Activator Transiently Enhances Blood–Brain Barrier Permeability During Cerebral Ischemia through Vascular Endothelial Growth Factor-Mediated Endothelial Endocytosis in Mice. J. Cereb. Blood Flow Metab. 2015, 35, 2021–2031.
- Suzuki, Y.; Nagai, N.; Umemura, K. A Review of the Mechanisms of Blood-Brain Barrier Permeability by Tissue-Type Plasminogen Activator Treatment for Cerebral Ischemia. Front. Cell. Neurosci. 2016, 10, 2.
- Rust, R.; Grönnert, L.; Weber, R.Z.; Mulders, G.; Schwab, M.E. Refueling the Ischemic CNS: Guidance Molecules for Vascular Repair. Trends Neurosci. 2019, 42, 644–656.
- Rust, R.; Weber, R.Z.; Grönnert, L.; Mulders, G.; Maurer, M.A.; Hofer, A.-S.; Sartori, A.M.; Schwab, M.E. Anti-Nogo-A antibodies prevent vascular leakage and act as pro-angiogenic factors following stroke. Sci. Rep. 2019, 9, 20040.