The Conventional Model of Colorectal Carcinogenesis and the “Serrated” Pathway
Colorectal cancer is a multifactorial and heterogeneous disease
[1]. Most CRCs (75%) are sporadic, whereas about 20% of CRC patients report a family history of the disease. Finally, 3–5% of CRCs are hereditary, with subjects bearing highly penetrant germline mutations that are associated with well-defined cancer-predisposing syndromes such as the hereditary nonpolyposis colorectal cancer (HNPCC), best known as Lynch syndrome (1–3%), or the familial adenomatous polyposis (FAP) (<1%), or again the hamartomatous polyposis syndrome, which displays the lowest incidence (<0.1%)
[2].
CRC pathogenesis is due to the progressive accumulation of genetic and epigenetic alterations, some of which being responsible for activating oncogenes or inactivating oncosuppressor genes, that are able to drive the malignant evolution from normal epithelium through early neoplastic lesions (aberrant crypt foci, adenomas, and serrated adenomas) to CRC
[3,4][3][4]. Such malignant transformation requires up to 15 years, depending on the characteristics of the lesion and on other independent risk factors such as gender, body weight, body mass index, physical inactivity
[5].
Neoplastic transformation affecting the colon epithelium is characterized by two distinct morphological pathways of carcinogenesis, namely the conventional and the alternative/serrated neoplasia pathways, each one being defined by specific genetic and epigenetic alterations, typical clinical and histological features and leading to different phenotypes
[6,7,8][6][7][8].
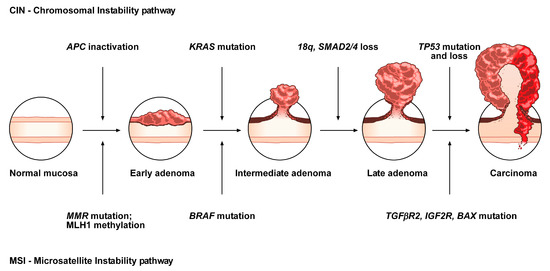
The conventional model, the so-called adenoma-carcinoma sequence, is histologically homogeneous and morphologically characterized the adenoma, including tubular or tubulovillous adenoma, as a precursor lesion
[1]. The adenoma-carcinoma sequence is a multistep mutational pathway, in which each histological alteration is the consequence of a molecular dysregulation
[9,10,11,12][9][10][11][12]. At the molecular level, this model recognizes a heterogeneous background, based on two mechanisms of tumorigenesis: (i) chromosomal instability (CIN) or (ii) microsatellite instability (MSI)
[13,14,15,16][13][14][15][16] (
Figure 1).
Figure 1. Conventional adenoma-to-carcinoma sequence. The chromosomal instability (CIN) pathway begins with bi-allelic mutations in the tumor suppressor gene APC within the normal colonic mucosa. The latter progressively differentiate into adenocarcinoma upon acquisition of additional mutations in the genes KRAS, SMAD4, and TP53, with consequent dysregulation of the Wnt/β-catenin, MAPK, PI3K and TGF-β signaling pathways. Alternatively, the MSI pathway involves an initial alteration of the Wnt signaling that leads to the formation of an early adenoma. Then, BRAF mutation followed by alterations of the genes TGFBR2, IGF2R, and BAX, participate in the progression toward the intermediate and late stages of carcinogenesis.
CIN represents the most prevalent form of genomic instability. It is detected in 85% of sporadic CRCs and is frequently observed in distal, rather than proximal, colon cancer sites
[15,16][15][16]. CIN consists of a gain or loss of all or part of chromosome(s), and is usually associated with mutations in proto-oncogenes or tumor suppressor genes, such as
KRAS or
APC, respectively
[13,16][13][16].
MSI occurs in about 15% of CRCs, predicts a favorable outcome in CRC and can also be detected in the serrated pathway
[15,17,18,19][15][17][18][19]. This genomic instability does not affect chromosomal integrity but consists of an accumulation of insertions/deletions of short nucleotide repeats (microsatellites) that is consecutive to hereditary (5%) or sporadic (10%) alterations in genes involved in DNA mismatch repair (MMR)
[14,15][14][15].
Although MSI, according to the National Cancer Institute, is frequently determined using a panel of five markers (BAT25, BAT26, D2S123, D5346, and D17S250), a variety of commercially available panels are currently used in most laboratories
[20]. Depending on the number of microsatellites associated with these markers, tumors have been subclassified into: (i) high, labeled “MSI”, (ii) low, labeled “MSI-L” or (iii) stable, labeled “MSS”
[21]. MSI-L tumors have been regrouped with MSS tumors, due to low differences in their clinicopathological characteristics or in most of their molecular features
[22].
Approximately, 3–15% of all CRCs are represented by sporadic forms with MSI
[21,23][21][23]. Several studies have demonstrated that epigenetic hypermethylation (80% of MSI CRCs), and the consequent silencing and inactivation of the gene
MLH1, is the event that triggers malignant transformation and determines a high rate of MSI
[21,23][21][23]. Moreover, mutations in MMR genes (20% of MSI CRCs) can also determine MSI tumors, associated with HNPCC (3% of CRCs)
[21,23][21][23]. HNPCC is an autosomal dominant disease due to germline mutations in some MMR genes (e.g.,
MSH2, MLH1, MSH6, PMS2, and
PMS1), causing consequent inactivation of the DNA repair system and the accumulation of mutated microsatellites
[24]. In addition, germline deletions in the 3’ end of
EPCAM result in epigenetic inactivation of the adjacent gene
MSH2 and represent another mutational mechanism responsible for HNPCC (1–3% of HNPCC patients)
[25]. HNPCC is not characterized by
MLH1 hypermethylation. Thus, MSI analysis, in addition to
MLH1 evaluation and
BRAF mutation analysis, is currently one of the first steps for the diagnosis of this disease
[24,26][24][26].
In contrast to the conventional adenoma-carcinoma pathway, an alternative pathway, featured by the presence of serrated adenomas/polyps as precursor lesions, has been documented over the last 10 years
[21,27,28,29,30,31][21][27][28][29][30][31]. It has been estimated that 15 to 30% of all CRCs arise from early neoplastic serrated lesions. These lesions, that are histologically characterized by a “serrated” (or saw-toothed) appearance of the epithelial glandular crypts within the precursor polyps, have long been considered innocuous
[31,32,33,34][31][32][33][34]. Nevertheless, serrated lesions are among the main causes of the “interval” CRCs and are associated with synchronous and metachronous advanced colorectal neoplasia
[35,36][35][36].
At the molecular level, serrated colorectal lesions rarely present truncating
APC mutations. The majority of CRCs arising from serrated lesions carry
BRAF mutations (whose prevalence varies among the different serrated subtypes), while
KRAS mutations remain less frequent. They are also associated with two pathways, namely MSI and the CpG island methylator phenotype (CIMP), which are involved in genomic instability; the latter being considered as the major mechanism that drives the serrated pathway toward CRC
[37,38][37][38].
Although the role of
APC mutations, and the subsequent aberrant activation of the WNT pathway, is fully understood in the conventional adenoma-carcinoma sequence, its role in the serrated pathway remains unclear. To address this issue, the mutational landscape of
APC in serrated precursors and
BRAF mutant cancers has been recently explored
[39]. In the cited study, even if the WNT pathway was notably activated in dysplastic serrated lesions and
BRAF mutant cancers, it was not due to truncating
APC mutations, suggesting the existence of alternative mechanisms of activation of the WNT signaling. Moreover, the role of missense
APC mutations, which are relatively frequent in serrated lesions and
BRAF mutant cancers with MSI, should be further investigated in the serrated pathway.
Overall, CRCs have been classified into five molecular subtypes based on their MSI and CIMP status, among which the three following signatures describe serrated lesions
[21]:
-
CIMP-H, MLH1 methylated, MSI, BRAF mutated lesions, known as sporadic MSI;
-
CIMP-H, MLH1 partially methylated, MSS, BRAF mutated lesions;
-
CIMP-L, MGMT methylated, MSS, KRAS mutated lesions.
2. Histopathological and Endoscopic Features of Serrated Colorectal Lesions
Serrated neoplasia of the colorectum represents one of the CRC subtypes
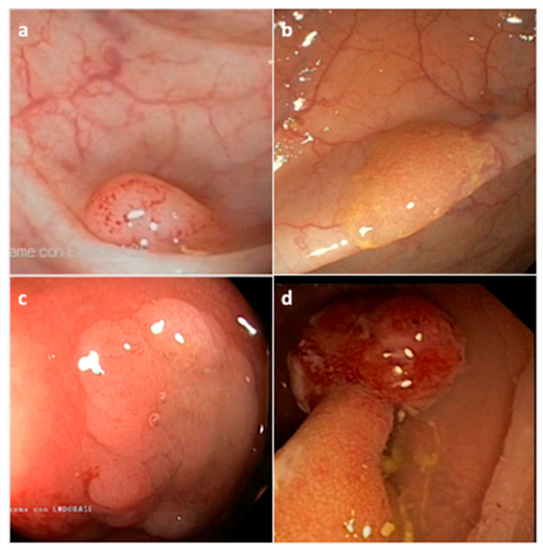
[45][40]. They are histologically classified by the World Health Organization (WHO) into three morphological categories: (i) hyperplastic polyp (HP), (ii) sessile serrated adenoma/polyp (SSA/P) with or without cytological dysplasia (SSAD), and (iii) the traditional serrated adenoma/polyp (TSA) (
Figure 2) (
Table 1)
[46][41]. The serrated subtypes, identified by their cytological characteristics and lesion area, have a distinct endoscopic appearance, share some histological features, and are unique at the biological and molecular levels
[34,47][34][42].
Figure 2. Representative endoscopic appearance of serrated lesions of the colorectum. (a) Hyperplastic polyp; (b) Sessile serrated adenoma/polyp; (c) Sessile serrated adenoma/polyp with dysplasia; (d) Traditional serrated adenoma. (Courtesy of Prof. Dr. Giovanni D. De Palma, University of Naples Federico II, Naples, Italy).
Table 1. Morphologic categories and features of serrated colorectal lesions.
| Histological Classification |
Frequency (%) * |
Location |
Shape |
Mucin Type |
Size |
| Hyperplastic polyp (HP) |
80–90% |
Distal |
Sessile, Flat |
Variable |
<5 mm |
| Microvesicular HP (MVHP) |
60% |
Distal |
Sessile |
Microvesicular |
<5 mm |
| Goblet cell HP (GCHP) |
30% |
Distal |
Sessile |
Goblet cells |
<5 mm |
| Mucin poor HP (MPHP) |
10% |
Distal |
Sessile |
Poor |
<5 mm |
| Sessile serrated adenoma/polyp (SSA/P) |
15–20% |
Proximal |
Sessile/Flat |
Microvesicular |
>5 mm |
| Traditional serrated polyp (TSA) |
1–6% |
Distal |
Sessile/Pedunculated |
Not present |
>5 mm |
However, alternative molecular classifications have been proposed based on recent findings, such as those defined by the CRC Subtyping Consortium (CRCSC) or by Fennell et al.
[40,41,42][50][51][52].
The heterogeneity of serrated lesions and the presence of morphological features shared with different subtypes, make difficult the accurate CRC classification during the diagnostic process and also the physiopathological interpretation of the observed lesions. In addition, serrated lesions with a distinctive endoscopic appearance are more difficult to detect compared to conventional lesions. In fact, detection of serrated lesions, particularly those located in the proximal colon, is difficult and endoscopist-dependent
[43,44][53][54]. Therefore, characterization of molecular markers specific for each CRC subtype may improve the identification of CRCs arising from this alternative pathway, and consequently support the diagnostic process as well as the clinical decision-making.
3. Molecular Features of the Serrated Colorectal Precursor Lesions
3.1. Hyperplastic Polyps
As described above, hyperplastic polyps can be histologically subclassified into MPHP, GCHP and MVHP lesions (
Table 1). The endoscopic diagnosis between these subtypes, and furthermore between HPs and SSA/Ps, is difficult and may be supported by the detection of specific biomarkers.
MPHP is the rarest form of HP and is not well described; in fact, to date, it has only been associated with CIMP-H (
Table 3). MVHP and GCHP are the most common HP subtypes. At the molecular level, MVHP is particularly characterized by
BRAF V600E mutation and CIMP-H (
Table 3); for that reason, it is considered a precursor of SSA/Ps
[17,82][17][55].
Table 3. Molecular profile of serrated colorectal lesions.
| Serrated Lesion |
BRAF/KRAS Status |
CIMP Rate |
Gene Methylation |
MSI Rate |
| HP |
BRAF mutated |
CIMP-H |
MLH1 not methylated |
MSS |
| MPHP * |
controversial |
CIMP-H |
controversial |
controversial |
| GCHP * |
KRAS mutated |
CIMP-L |
MLH1 not methylated |
MSS |
| MVHP * |
BRAF mutated |
CIMP-H |
MLH1 not methylated |
MSS |
| SSA/P |
BRAF mutated |
CIMP-H |
MLH1 not methylated |
MSS |
| SSAD |
BRAF mutated |
CIMP-H |
MLH1 hypermethylated |
MSI |
| TSA |
KRAS/BRAF mutated or neither |
CIMP-L/-H |
MLH1 not methylated |
MSS |
| TSA HGD |
KRAS mutated |
CIMP-L |
MGMT hypermethylated |
MSS |