Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Peter Tang and Version 3 by Dean Liu.
Trimethylamine-N-oxide (TMAO) is one among a group of selective uremic toxins that may rise to high levels in the circulation or accumulate in various organs. Diet and microbiota each have a direct impact on many chronic, inflammatory, and metabolic diseases. As the field develops, a new perspective is emerging. The effects of diet may depend on the microbiota composition of the intestine. A diet that is rich in choline, red meat, dairy, or egg may promote the growth, or change the composition, of microbial species.
- trimethylamine-N-oxide
- cardiovascular disease
- rheumatoid arthritis
1. Introduction
Recent guidelines from the European League Against Rheumatism (EULAR) recommend aggressive management of cardiovascular disease (CVD) risk factors in addition to rheumatoid arthritis (RA) disease activity control, because severe subclinical atherosclerotic diseases underlie rheumatoid arthritis. Thirty to fifty percent of RA patients’ life expectancy is shorter than normal by 5–10 years, with heart attacks and strokes as the major causes of death.
Diet has always been an essential factor in the management of atherosclerosis, RA, and other autoimmune diseases. More recently, the impact of dysbiosis, lack of diversity or imbalance of the intestinal microbiota has been recognized as a crucial factor in autoimmunity. Emerging from these two fields is the discovery that certain groups of intestinal bacteria process dietary compounds to produce metabolites that may benefit or compromise the host’s homeostasis [1].
2. Microbiome Alters the Dietary Composition
The human body, especially the intestine, is colonized by a vast amount of microbes. These intestinal bacteria, comprised of trillions of typically nonpathogenic commensal organisms, weigh nearly 0.2 kg [2][3][4]. The most abundant bacterial genera in the healthy gut are from the phylum Firmicutes, and within it, the most significant classes are the Gram-positive Clostridia and the Gram-negative Bacteroides. The Gram-negative Proteobacteria, with the Gram-positive Actinobacteria, Gram-negative Fusobacteria, and Verrucomicrobia, is also notable within the gut flora.
Human intestinal bacteria produce a variety of metabolites, some that benefit and others that compromise the host’s homeostasis [1]. The primary metabolites are branched-chain amino acids (BCAAs) and short-chain fatty acids (SCFAs). SCFAs are derived from bacterial fermentation of dietary carbohydrates and are used for energy and nutrients. The major SCFAs are acetate, propionate, and butyrate. Muscle uses acetic acid and the liver uses propionic acid to facilitate ATP production. Butyric acid provides energy to gut cells [5] and promotes the regulatory T lymphocytes, which calm the mucosal immune response, suppress inflammation, and maintain mucosal integrity in the colon [6]. Many enteric bacteria in the phyla Firmicutes, Bacteroidetes, and Actinobacteria, which encompass the probiotics in the orders of Bifidobacteriales and Lactobacillales, produce them [1]. BCAAs are derivatives from essential amino acids. Clostridium sporogenes metabolizes tryptophan to indole and subsequently to 3-indolepropionic acid, a highly potent neuroprotective antioxidant that beneficially scavenges hydroxyl radicals [6][7]. In contrast, Prevotella copri and Bacteroides vulgatus produce BCAAs that increase various health risks, for example, insulin resistance in humans [8].
TMAO is a microbiota-generated metabolite derived from choline and carnitine, which are essential nutrients contained in many foods, including red meat, eggs, and dairy. Choline has a wide range of biological activities; it maintains the structural integrity of cell membranes, supports cholinergic neurotransmission, and donates methyl groups in many biosynthetic reactions [9]. Carnitine transports long-chain fatty acids into the mitochondria to produce energy. The conversion of choline and carnitine to TMAO depends on the balance and diversity of the microbiota.
In animal models, Chen et al. (2016) detected a higher number of Prevotella and a lower number of Bacteroides, Lactobacillus, and Bifidobacterium within the fecal content of mice fed choline [10]. Bacteria in the human gut possess the trimethylamine lyase system (CutC/D) and the carnitine Rieske-type oxygenase/reductase system (CntA/B and YeaW/X) for metabolizing choline and carnitine in the diet into trimethylamine (TMA). In the human liver, TMA is oxidized by flavin-containing monooxygenases (FMO) to TMAO [11][12] (Figure 1). Romano et al. (2015) identified by metabolomics eight distinct bacterial strains of TMA producers: Anaerococcus hydrongenalis, Clostridium asparagiforme, Clostridium hathewayi, Clostridium sporogenes, Edwardsiella tarda, Escherichia fergusonii, Proteus penneri, and Providencia rettgeri [13].
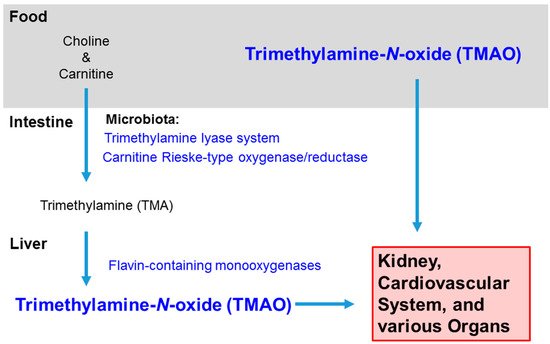
Figure 1. Sources and Distribution of Trimethylamine-N-oxide. TMAO intake can be direct, such as from fish, or indirect, converted from choline and carnitine in red meat, eggs, dairy, etc. In the intestine, bacteria in the microbiota convert choline and carnitine to trimethylamine (TMA), and then the hepatic enzymes flavin-containing monooxygenases further metabolize the molecule to trimethylamine-N-oxide. TMAO usually leaves the body in the urine, but it can accumulate in the kidney and rise to high levels in the circulation and various organs with inflammation.
3. TMAO Promotes the Etiological Mechanisms of CVD in RA
TMAO exerts an impact on many major mechanisms in the atherosclerosis pathogenesis pathway. It promotes forward cholesterol transport and inhibits reverse cholesterol transport (RCT). It alters the function of macrophages, foam cells, endothelial cells, and the responsiveness of platelets [11].
Atherosclerosis initiates with hypercholesterolemia. TMAO interferes with the elimination of cholesterol. RCT is a crucial process that uses high-density lipoproteins (HDLs) to move excess cholesterol from peripheral tissues to the liver where the bile would convert the cholesterol into bile acids for excretion in the feces [11]. TMAO impedes this pathway by interrupting bile synthesis and metabolism [10][14]. TMAO downregulates two cytochrome P450 enzymes, cholesterol 7 a-hydroxylase (cyp7A1) that is rate-limiting for bile synthesis, and sterol 27-hydroxylase that participates in diverting cholesterol into bile acids. Hence, it decreases recirculation into the intestine for absorption [15]. Ding et al. (2018) showed that TMAO significantly lowers the total bile acid pool size in mice [14]. TMAO supplementation also markedly reduces the expression of two intestinal cholesterol transporters—Niemann-Pick C1 Like1 (Npc1L1), which transports cholesterol into enterocytes from the gut lumen, and ATP-binding cassette subfamily G members 5 and 8 (abcg5/8) which transport cholesterol from the hepatobiliary [16][17].
Elevated TMAO levels in serum affect the influx/efflux of fatty deposits on the artery wall and set the stage for atherosclerosis [18]. The accumulated cholesterol, carried on low-density lipoprotein (LDL) and very low-density lipoprotein (VLDL), penetrates the endothelium. Arterial macrophages infiltrate the subendothelial spaces to ingest the LDL and VLDL. Macrophages take up lipids with their scavenger receptors CD-36 and scavenger receptor A (SRA). Typically, they limit lipid accumulation, digesting the LDL into fatty acids, free or esterified cholesterol to excrete via the apolipoprotein A1 (ApoA1) component of HDL. TMAO, however, disrupts the homeostatic mechanisms by causing an imbalance in the uptake and efflux with the upregulation of CD-36 and SRA receptors [19]. In combination with impaired RCT by bile, cholesterol accumulates within the macrophages [20]. Many clinical studies have revealed a direct correlation between the plasma levels of TMAO with the burden and extent of coronary atherosclerotic plaque [9][21][22][23].
The fat-laden macrophages and smooth muscle cells become the foam cells that contribute to inflammation of the arteries and lead to atherosclerosis. They produce reactive oxygen species (ROS), proteases, chemokines, pro-inflammatory cytokines, and eicosanoids, leading to vessel dilation, damage, thrombosis, and hypoxia [20]. TMAO further enhances oxidative stress by inhibiting manganese superoxide dismutase 2 activation and sirtuin 3 expression, so the ROS are not effectively neutralized, as shown in human umbilical vein endothelial cells (HUVECs) [24][25]. In a redox-regulated manner, TMAO (30 µM) activates the nucleotide-binding oligomerization domain-like receptor family pyrin domain containing 3 (NLRP3) inflammasome, caspase-1 activity, and IL-1β and IL-18 production [25][26][27]. Boini et al. (2017) showed the ROS-thioredoxin-interacting protein (TXNIP)-NLRP3 pathway mediates the inflammatory actions of TMAO [26]. The ROS inhibitor N-acetylcysteine and mitochondrial ROS scavenger process can neutralize its effect [27][28].
Hypertension is a recognized cause of cardiovascular and CKD, with the latter being another significant TMAO health risk. The peptide hormone angiotensin is widely employed in blood pressure studies for it regulates blood pressure and promotes sodium retention by the kidneys. In a study with Sprague–Dawley rats, co-infusion of TMAO with angiotensin II induced hypertension at 100-fold above normal levels, even though TMAO did not affect blood pressure in normal, nonhypertensive animals. It sustained the arterial pressure in the angiotensin-induced hypertensive rats, prolonging it two-fold, from 5 days to 14 days. Both the systolic and diastolic blood pressure increased compared to the rats who had angiotensin infusion alone [29].
Endothelial dysfunction is an early event in atherosclerosis. Li et al. (2017; 2018) revealed the effect of TMAO on endothelial nitric oxide synthase (eNOS) using rats in a CKD model [30][31]. Normal 22-month-old rats had low levels of TMAO (14 µM) whereas those with CKD were 3–4 fold higher (50 µM), and their aorta had suppressed acetylcholine-dependent endothelium relaxation and eNOS activity. These suppressions are reversible by adding an inhibitor of trimethylamine formation, 3,3-dimethyl-1-butanol, in the drinking water [30][31]. In vitro, TMAO (30 µM) increases the loss in interendothelial tight junction integrity. A decrease in tight junction protein zonula occludens-1 (ZO-1) and an increase in dextran flux in cultured mouse carotid arterial endothelial cells reflect the increase in vascular permeability [26]. TMAO (100 µM) also downregulates the production of nitric oxide and increases the mRNA and protein expression of the intercellular adhesion molecule (ICAM) and the vascular cell adhesion protein (VCAM) in primary HUVECs [28][32].
TMAO increases the incidence of thrombotic events, such as heart attacks and strokes [33]. Platelets move to the site of vascular injury to form plugs and enter a state of heightened reactivity. The activated platelets release various vasoactive compounds, clotting factors, and cytokines; then thrombosis, strokes, and myocardial infarctions occur. Evidence from humans and mice suggests that TMAO promotes platelet aggregation [33]. A study involving over 4000 subjects has shown that plasma levels of TMAO predict the incidence of thrombotic events [33]. Cheng et al. (2019) showed that TMAO promotes tissue factor activity in tumor necrosis factor-stimulated primary human coronary aortic endothelial cells. They also reported that the plasma TMAO level positively correlates with tissue factor activity in patients with ST-elevation myocardial infarction (STEMI) [34]. Animal models provide evidence that TMAO is causal to thrombosis [12]. Using antisense oligonucleotide targeting suppression and transgene, overexpression of the TMAO-generating enzyme, flavin-containing monooxygenase (FMO), Zhu et al. (2018) showed that plasma TMAO levels could modulate platelet activity [35] (Figure 2).
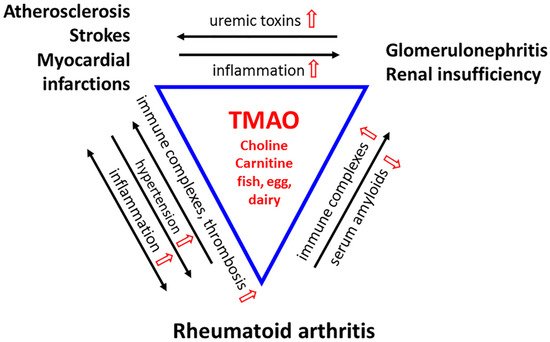
Figure 2. A Triad of Three Diseases. Rheumatoid arthritis (RA) is a chronic inflammatory disease in which immune complexes (RF and ACPA) induce inflammation in the microvasculature of the synovial joint. The stress is systemic. Cytokines produced locally disseminate into the circulatory system to cause inflammation in the arteries and veins. Chronic vascular inflammation leads to hypertension and hypoxia, which facilitates the development of RA-fibroblast-like synoviocytes (FLS) that invade the bone. Inflammatory stress leads to endothelial dysfunction, deactivation of endothelial nitric oxide synthase (eNOS), and compromised drainage. The increase in pro-inflammatory cytokines, chemokines, and adhesion molecules promotes the formation of atherosclerotic vascular lesions. These reactions feedback to the joints, exacerbating their inflammation and causing injury to the microvasculature. Platelets are hyperactivated, and unstable plagues increase the incidence of thrombosis, heart attacks, and strokes, the significant causes of death in RA patients. The immune complexes deposit in the glomeruli membrane, activating complement to damage the kidney. Uremic toxin levels rise throughout the circulation. Renal insufficiency promotes atherosclerosis through inflammation and vice versa. TMAO, derived from diet, heightens all these inflammatory reactions and leads to perpetuating cycles. Nonetheless, as a chemical chaperone and osmolyte, TMAO would protect proteins from the toxicity of urea, and acute phase proteins from aggregating into amyloids that compromise renal functions. (TMAO and its modulations are in red).
4. Microbial Dysbiosis and TMAO Promote Rheumatoid Arthritis
In addition to metabolic diseases, microbial dysbiosis plays a role in the pathogenesis of autoimmune diseases both within and outside of the gastrointestinal (GI) tract [36]. Rheumatoid arthritis (RA) is an example of these autoimmune diseases. It affects 1.3 million Americans [37][38]. RA patients harbor the TMAO-producing anaerobic, Gram-negative Bacteriodetes, Prevotella copri. It is abundant in the periodontium, the lung, and the intestine, correlating with adverse cardiovascular events in RA patients [39][40]. Furthermore, it may alter the metabolism of the microbiota to reduce the effectiveness of the common disease-modifying antirheumatic drug (DMARD) methotrexate [41].
The most noticeable symptoms of RA are inflammation of the synovial tissues and erosion of cartilage and bone in the joints of hands and feet. Nonetheless, RA is a systemic disease with cardiovascular risk factors, which include hypertension, hyperlipidemia, vasculitis, atherosclerosis, and endothelial dysfunction. RA patients may harbor severe subclinical atherosclerotic diseases and have up to a three-fold increased risk of cardiovascular disease. Recent clinical studies discovered that atherosclerosis can lead to a shortened life expectancy in RA patients. Fatal heart attacks and strokes are the significant causes of mortality in these individuals [42][43][44]. Okano et al. (2017) and Olah et al. (2017) detected that carotid atherosclerosis plaque with distal occlusion in the middle cerebral artery (MCA) are more frequent and severe in RA patients [45][46]. The pronounced distal occlusions impair circulatory reserve capacity. CVD is a formidable risk in RA patients [47].
Collagen-induced arthritis (CIA) is one of the most widely accepted murine models for human RA. The DBA/1 strain of mice used in this model is uniquely susceptible to developing RA in response to immunization with type II collagen (CII) plus complete Freund’s adjuvant (CFA) [48][49][50][51]. Interestingly, these mice are also susceptibility to dyslipidemia when on an atherogenic diet (https://www.jax.org/strain/000670) [52][53]. After a high-fat diet for 14 weeks, there are both dyslipidemia and atherosclerotic lesions [54].
RA and atherosclerosis share many pathological mechanisms as extensively shown in humans and rodent models. Mice develop severe arthritis in joints from anti-collagen or anti-glucose-6-phosphate isomerase (G6PI) antibodies [49][55][56]. RA patients develop autoantibodies such as rheumatoid factor (IgM antibody to self IgG) and anti-citrullinated peptide antibody (ACPA) [57][58][59]. Citrullinated fibrinogen, citrullinated vimentin, and citrullinated histone H2B are present within both the atherosclerotic plaques and the arthritic joints of patients [60][61][62]. Whereas high titers of ACPA are in the circulation of RA patients [46], ACPA, such as anti-citrullinated vimentin, are in the calcified coronary arteries [46][63]. The citrullination and ACPAs could be the underlying mechanism for accelerated atherosclerosis and severe carotid atherosclerotic plaque in RA patients. This is because the autoantibodies form immune complexes that stimulate the inflammatory process and cause tissue damage via the complement system [64]. Continuous production of such immune complexes ultimately results in the chronic inflammation that is characteristic for RA [65] (Figure 2).
5. TMAO Can Harm or Protect the Renal Function of RA Patients
RA patients, as well as many who suffer from other rheumatic diseases, are at risk for renal insufficiency [66][67][68]. Approximately 15–25% of RA patients have reduced glomerular filtration rate (GFR) <60 mL/min [69], even though the frequency of end-stage renal disease in patients with RA is 1% [67]. The nephrotoxicity of therapeutic drugs, such as NSAIDs, d-penicillamine, and gold, are among the culprits that cause proteinuria and the concurrent renal disease that significantly increase mortality in RA patients.
Even though TMAO and its probable signaling mechanisms are associated with CVD and CKD, a contention exists regarding whether it would harm or protect the renal system. In RA, IL-6 activates the production of acute phase proteins, which can aggregate into toxic insoluble serum amyloid A. The amyloids deposit in the glomeruli, blood vessels, peritubular, and interstitium, resulting in kidney damage. CKD patients classically present with proteinuria, increase in serum creatinine level, and then progressive renal failure. Renal biopsies from RA patients with a clinical renal disease showed that 30% have amyloidosis, and 17% have membranous glomerulonephritis [66].
TMAO may critically benefit renal function in systemic inflammatory diseases by being a “chemical chaperone” to enhance the folding and stability of proteins, and an osmolyte to maintain cellular fluid balance [70][71]. It accumulates in the endoplasmic reticulum to promote protein folding, thereby inhibiting ER stress and attenuating the formation of intracellular aggregates [72]. In the kidneys, accumulated TMAO counteracts the destabilizing effects of urea on macromolecular structures of proteins and nucleic acids, and offsets the inhibitory effects of urea on functions such as ligand binding [72].
The amyloids can also accumulate and aggregate in other tissues and organs, causing cardiomyopathy, hypertension, and corneal and vitreous abnormalities. TMAO protects epidermolysis bullosa simplex keratinocytes from heat stress-induced keratin aggregation. This suggests a therapeutic potential for TMAO in epidermolysis bullosa and other keratinopathies [73]. TMAO modulates binding of protein kinase A-phosphorylated tau protein to tubulin, implicating a therapeutic application in Alzheimer’s disease [71][74]. There is also a study that shows a moderate increase in plasma TMAO does not harm the circulatory system. In contrast, increased dietary TMAO seems to reduce diastolic dysfunction in the pressure-overloaded heart in spontaneously hypertensive rats. Therefore, TMAO may be renal protective in RA patients (Figure 2).
6. Microbiota Remodeling Reverses TMAO-Mediated Pathologies
Although TMAO at high levels poses risks for metabolic and autoimmune diseases in humans, many studies have shown that its levels may be modulated via remodeling the gut microbiota. For example, consumption of phytochemicals, such as allicin in garlic and resveratrol in red wine and grapes, may change the composition of the gut microbiota and, in association, reduce the effect of TMAO. Wu et al. (2015) examined the effect of allicin (50 mg) on carnitine diet using C57Bl/6 mice [75]. Oral feeding of allicin reduced the carnitine-induced plasma level of TMAO to that of standard chow, concomitant with a change in the taxa of the gut microbiota. Similarly, Chen et al. (2016) have observed remodeling and inhibition of TMAO synthesis in C57BL/6 mice and the apolipoprotein E-deficient mice fed choline and treated with resveratrol [10]. When on a choline diet, the mice had an increased number of Prevotella, a decreased number of Lactobacillus and Bifidobacteria, and increased TMAO levels. Upon treatment with resveratrol (0.4% of chow), the microbial population levels reversed and the plasma TMAO levels decreased.
References
- Louis, P.; Flint, H.J. Formation of Propionate and Butyrate by the Human Colonic Microbiota. Environ. Microbiol. 2017, 19, 29–41.
- Bäckhed, F. Host-Bacterial Mutualism in the Human Intestine. Science 2005, 307, 1915–1920.
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533.
- Costea, P.I.; Hildebrand, F.; Arumugam, M.; Bäckhed, F.; Blaser, M.J.; Bushman, F.D.; de Vos, W.M.; Ehrlich, S.D.; Fraser, C.M.; Hattori, M.; et al. Enterotypes in the Landscape of Gut Microbial Community Composition. Nat. Microbiol. 2018, 3, 8–16.
- Gibson, G.R. Fibre and effects on probiotics (the prebiotic concept). Clin. Nutr. Suppl. 2004, 1, 25–31.
- Honda, K.; Littman, D.R. The microbiota in adaptive immune homeostasis and disease. Nature 2016, 535, 75–84.
- Chyan, Y.J.; Poeggeler, B.; Omar, R.A.; Chain, D.G.; Frangione, B.; Ghiso, J.; Pappolla, M.A. Potent neuroprotective properties against the Alzheimer beta-amyloid by an endogenous melatonin-related indole structure, indole-3-propionic acid. J. Biol. Chem. 1999, 274, 21937–21942.
- Zhao, X.; Han, Q.; Liu, Y.; Sun, C.; Gang, X.; Wang, G. The Relationship between Branched-Chain Amino Acid Related Metabolomic Signature and Insulin Resistance: A Systematic Review. J. Diabetes Res. 2016, 2016, 2794591.
- Tang, W.H.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal Microbial Metabolism of Phosphatidylcholine and Cardiovascular Risk. N. Engl. J. Med. 2013, 368, 1575–1584.
- Chen, M.-L.; Yi, L.; Zhang, Y.; Zhou, X.; Ran, L.; Yang, J.; Zhu, J.-D.; Zhang, Q.-Y.; Mi, M.-T. Resveratrol Attenuates Trimethylamine-N-Oxide (TMAO)-Induced Atherosclerosis by Regulating TMAO Synthesis and Bile Acid Metabolism via Remodeling of the Gut Microbiota. mBio 2016, 7, e02210-15.
- Canyelles, M.; Tondo, M.; Cedó, L.; Farràs, M.; Escolà-Gil, J.C.; Blanco-Vaca, F. Trimethylamine N-Oxide: A Link among Diet, Gut Microbiota, Gene Regulation of Liver and Intestine Cholesterol Homeostasis and HDL Function. Int. J. Mol. Sci. 2018, 19, 3228.
- Koeth, R.A.; Lam-Galvez, B.R.; Kirsop, J.; Wang, Z.; Levison, B.S.; Gu, X.; Copeland, M.F.; Bartlett, D.; Cody, D.B.; Dai, H.J.; et al. L-Carnitine in Omnivorous Diets Induces an Atherogenic Gut Microbial Pathway in Humans. J. Clin. Investig. 2019, 129, 373–387.
- Romano, K.A.; Vivas, E.I.; Amador-Noguez, D.; Rey, F.E. Intestinal Microbiota Composition Modulates Choline Bioavailability from Diet and Accumulation of the Proatherogenic Metabolite Trimethylamine-N-Oxide. mBio 2015, 6, e02481-14.
- Ding, L.; Chang, M.; Guo, Y.; Zhang, L.; Xue, C.; Yanagita, T.; Zhang, T.; Wang, Y. Trimethylamine-N-oxide (TMAO)-induced atherosclerosis is associated with bile acid metabolism. Lipids Health Dis. 2018, 17, 286.
- Huang, F.; Zhang, F.; Xu, D.; Zhang, Z.; Xu, F.; Tao, X.; Qiu, L.; Wei, H. Enterococcus faecium WEFA23 from infants lessens high-fat-diet-induced hyperlipidemia via cholesterol 7-alpha-hydroxylase gene by altering the composition of gut microbiota in rats. J. Dairy Sci. 2018, 101, 7757–7767.
- Liu, S.; You, L.; Zhao, Y.; Chang, X. Wild Lonicera caerulea berry polyphenol extract reduces cholesterol accumulation and enhances antioxidant capacity in vitro and in vivo. Food Res. Int. 2018, 107, 73–83.
- Mohammadi, A.; Najar, A.G.; Yaghoobi, M.M.; Jahani, Y.; Vahabzadeh, Z. Trimethylamine-N-Oxide Treatment Induces Changes in the ATP-Binding Cassette Transporter A1 and Scavenger Receptor A1 in Murine Macrophage J774A.1 Cells. Inflammation 2016, 39, 393–404.
- Mondal, J.A. Effect of Trimethylamine N-Oxide on Interfacial Electrostatics at Phospholipid Monolayer—Water Interfaces and Its Relevance to Cardiovascular Disease. J. Phys. Chem. Lett. 2016, 7, 1704–1708.
- Geng, J.; Yang, C.; Wang, B.; Zhang, X.; Hu, T.; Gu, Y.; Li, J. Trimethylamine N-oxide promotes atherosclerosis via CD36-dependent MAPK/JNK pathway. Biomed. Pharmacother. 2018, 97, 941–947.
- Haghikia, A.; Li, X.S.; Liman, T.G.; Bledau, N.; Schmidt, D.; Zimmermann, F.; Kränkel, N.; Widera, C.; Sonnenschein, K.; Haghikia, A.; et al. Gut Microbiota-Dependent Trimethylamine N-Oxide Predicts Risk of Cardiovascular Events in Patients with Stroke and Is Related to Proinflammatory Monocytes. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2225–2235.
- Bennett, B.J.; Davis, R.C.; Civelek, M.; Orozco, L.; Wu, J.; Qi, H.; Pan, C.; Packard, R.R.S.; Eskin, E.; Yan, M.; et al. Genetic Architecture of Atherosclerosis in Mice: A Systems Genetics Analysis of Common Inbred Strains. PLoS Genet. 2015, 11, e1005711.
- Liu, X.; Xie, Z.; Sun, M.; Wang, X.; Li, J.; Cui, J.; Zhang, F.; Yin, L.; Huang, D.; Hou, J.; et al. Plasma trimethylamine N-oxide is associated with vulnerable plaque characteristics in CAD patients as assessed by optical coherence tomography. Int. J. Cardiol. 2018, 265, 18–23.
- Kiouptsi, K.; Reinhardt, C. Contribution of the commensal microbiota to atherosclerosis and arterial thrombosis. Br. J. Pharmacol. 2018, 175, 4439–4449.
- Sun, G.; Yin, Z.; Liu, N.; Bian, X.; Yu, R.; Su, X.; Zhang, B.; Wang, Y. Gut microbial metabolite TMAO contributes to renal dysfunction in a mouse model of diet-induced obesity. Biochem. Biophys. Res. Commun. 2017, 493, 964–970.
- Chen, M.; Zhu, X.; Ran, L.; Lang, H.; Yi, L.; Mi, M. Trimethylamine-N-Oxide Induces Vascular Inflammation by Activating the NLRP3 Inflammasome Through the SIRT3-SOD2-mtROS Signaling Pathway. J. Am. Heart Assoc. 2017, 6, e006347.
- Boini, K.M.; Hussain, T.; Li, P.-L.; Koka, S. Trimethylamine-N-Oxide Instigates NLRP3 Inflammasome Activation and Endothelial Dysfunction. Cell. Physiol. Biochem. 2017, 44, 152–162.
- Ke, Y.; Li, D.; Zhao, M.; Liu, C.; Liu, J.; Zeng, A.; Shi, X.; Cheng, S.; Pan, B.; Zheng, L.; et al. Gut flora-dependent metabolite Trimethylamine-N-oxide accelerates endothelial cell senescence and vascular aging through oxidative stress. Free Radic. Biol. Med. 2018, 116, 88–100.
- Sun, X.; Jiao, X.; Ma, Y.; Liu, Y.; Zhang, L.; He, Y.; Chen, Y. Trimethylamine N-oxide induces inflammation and endothelial dysfunction in human umbilical vein endothelial cells via activating ROS-TXNIP-NLRP3 inflammasome. Biochem. Biophys. Res. Commun. 2016, 481, 63–70.
- Ufnal, M.; Jazwiec, R.; Dadlez, M.; Drapala, A.; Sikora, M.; Skrzypecki, J. Trimethylamine-N-Oxide: A Carnitine-Derived Metabolite That Prolongs the Hypertensive Effect of Angiotensin II in Rats. Can. J. Cardiol. 2014, 30, 1700–1705.
- Li, T.; Chen, Y.; Gua, C.; Li, X. Elevated Circulating Trimethylamine N-Oxide Levels Contribute to Endothelial Dysfunction in Aged Rats through Vascular Inflammation and Oxidative Stress. Front. Physiol. 2017, 8, 350.
- Li, T.; Gua, C.; Wu, B.; Chen, Y. Increased circulating trimethylamine N-oxide contributes to endothelial dysfunction in a rat model of chronic kidney disease. Biochem. Biophys. Res. Commun. 2018, 495, 2071–2077.
- Ma, G.; Pan, B.; Chen, Y.; Guo, C.; Zhao, M.; Zheng, L.; Chen, B. Trimethylamine N-oxide in atherogenesis: Impairing endothelial self-repair capacity and enhancing monocyte adhesion. Biosci. Rep. 2017, 37.
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell 2016, 165, 111–124.
- Cheng, X.; Qiu, X.; Liu, Y.; Yuan, C.; Yang, X. Trimethylamine N-oxide promotes tissue factor expression and activity in vascular endothelial cells: A new link between trimethylamine N-oxide and atherosclerotic thrombosis. Thromb. Res. 2019, 177, 110–116.
- Zhu, W.; Buffa, J.A.; Wang, Z.; Warrier, M.; Schugar, R.; Shih, D.M.; Gupta, N.; Gregory, J.C.; Org, E.; Fu, X.; et al. Flavin monooxygenase 3, the host hepatic enzyme in the metaorganismal trimethylamine N-oxide-generating pathway, modulates platelet responsiveness and thrombosis risk. J. Thromb. Haemost. 2018, 16, 1857–1872.
- Brusca, S.B.; Abramson, S.B.; Scher, J.U. Microbiome and mucosal inflammation as extra-articular triggers for rheumatoid arthritis and autoimmunity. Curr. Opin. Rheumatol. 2014, 26, 101–107.
- Helmick, C.G.; Felson, D.T.; Lawrence, R.C.; Gabriel, S.; Hirsch, R.; Kwoh, C.K.; Liang, M.H.; Kremers, H.M.; Mayes, M.D.; Merkel, P.A.; et al. Estimates of the Prevalence of Arthritis and Other Rheumatic Conditions in the United States. Arthritis Rheum. 2008, 58, 15–25.
- National Collaborating Centre for Chronic Conditions (UK). Rheumatoid Arthritis: National Clinical Guideline for Management and Treatment in Adults; Royal College of Physicians: London, UK, 2009.
- Scher, J.U.; Sczesnak, A.; Longman, R.S.; Segata, N.; Ubeda, C.; Bielski, C.; Rostron, T.; Cerundolo, V.; Pamer, E.G.; Abramson, S.B.; et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. eLife 2013, 2, e01202.
- Vaahtovuo, J.; Munukka, E.; Korkeamäki, M.; Luukkainen, R.; Toivanen, P. Fecal microbiota in early rheumatoid arthritis. J. Rheumatol. 2008, 35, 1500–1505.
- Chung, Y.-L.; Rider, L.G.; Bell, J.D.; Summers, R.M.; Zemel, L.S.; Rennebohm, R.M.; Passo, M.H.; Hicks, J.; Miller, F.W.; Scott, D.L.; et al. Muscle metabolites, detected in urine by proton spectroscopy, correlate with disease damage in juvenile idiopathic inflammatory myopathies. Arthritis Rheum. 2005, 53, 565–570.
- Blum, A.; Adawi, M. Rheumatoid arthritis (RA) and cardiovascular disease. Autoimmun. Rev. 2019, 18, 679–690.
- Rawla, P. Cardiac and vascular complications in rheumatoid arthritis. Reumatologia 2019, 57, 27–36.
- Gualtierotti, R.; Ughi, N.; Marfia, G.; Ingegnoli, F. Practical Management of Cardiovascular Comorbidities in Rheumatoid Arthritis. Rheumatol. Ther. 2017, 4, 293–308.
- Oláh, C.; Kardos, Z.; Sepsi, M.; Sas, A.; Kostyál, L.; Bhattoa, H.P.; Hodosi, K.; Kerekes, G.; Tamási, L.; Valikovics, A.; et al. Assessment of intracranial vessels in association with carotid atherosclerosis and brain vascular lesions in rheumatoid arthritis. Arthritis Res. Ther. 2017, 19, 213.
- Okano, T.; Inui, K.; Sugioka, Y.; Sugioka, K.; Matsumura, Y.; Takahashi, S.; Tada, M.; Mamoto, K.; Wakitani, S.; Koike, T.; et al. High titer of anti-citrullinated peptide antibody is a risk factor for severe carotid atherosclerotic plaque in patients with rheumatoid arthritis: The TOMORROW study. Int. J. Rheum. Dis. 2017, 20, 949–959.
- Jagpal, A.; Navarro-Millán, I. Cardiovascular co-morbidity in patients with rheumatoid arthritis: A narrative review of risk factors, cardiovascular risk assessment and treatment. BMC Rheumatol. 2018, 2, 10.
- Kato, F.; Nomura, M.; Nakamura, K. Arthritis in mice induced by a single immunisation with collagen. Ann. Rheum. Dis. 1996, 55, 535–539.
- Myers, L.K.; Rosloniec, E.F.; Cremer, M.A.; Kang, A.H. Collagen-induced arthritis, an animal model of autoimmunity. Life Sci. 1997, 61, 1861–1878.
- Jones, G.W.; Hill, D.G.; Sime, K.; Williams, A.S. In Vivo Models for Inflammatory Arthritis. Methods Mol. Biol. 2018, 1725, 101–118.
- Vincent, T.L.; Williams, R.O.; Maciewicz, R.; Silman, A.; Garside, P.; Arthritis Research UK animal Models Working Group. Mapping Pathogenesis of Arthritis through Small Animal Models. Rheumatology 2012, 51, 1931–1941.
- Roberts, A.; Thompson, J.S. Genetic factors in the development of atheroma and on serum total cholesterol levels in inbred mice and their hybrids. Prog. Biochem. Pharmacol. 1977, 13, 298–305.
- Roberts, A.; Thompson, J.S. Inbred mice and their hypbrids as an animal model for atherosclerosis research. Adv. Exp. Med. Biol. 1976, 67, 313–327.
- Paigen, B.; Morrow, A.; Brandon, C.; Mitchell, D.; Holmes, P. Variation in susceptibility to atherosclerosis among inbred strains of mice. Atherosclerosis 1985, 57, 65–73.
- Svetlicky, N.; Kivity, S.; Odeh, Q.; Shovman, O.; Gertel, S.; Amital, H.; Gendelman, O.; Volkov, A.; Barshack, I.; Bar-Meir, E.; et al. Anti-citrullinated-protein-antibody-specific intravenous immunoglobulin attenuates collagen-induced arthritis in mice. Clin. Exp. Immunol. 2015, 182, 241–250.
- Monach, P.A.; Mathis, D.; Benoist, C. The K/BxN Arthritis Model. Curr. Protoc. Immunol. 2008, 81, 15.22.1–15.22.12.
- Schoels, M.; Bombardier, C.; Aletaha, D. Diagnostic and Prognostic Value of Antibodies and Soluble Biomarkers in Undifferentiated Peripheral Inflammatory Arthritis: A Systematic Review. J. Rheumatol. Suppl. 2011, 87, 20–25.
- Lin, K.M.; Chen, W.M.; Tung, S.Y.; Wei, K.L.; Shen, C.H.; Chang, T.S.; Chang, P.J. Prevalence and Predictive Value of High-Positive Rheumatoid Factor and Anti-Citrullinated Protein Antibody Levels in Nonarthritic Patients with Chronic Hepatitis C Infection. Int. J. Rheum. Dis. 2019, 22, 116–120.
- Vossenaar, E.R.; Van Venrooij, W.J. Citrullinated proteins: Sparks that may ignite the fire in rheumatoid arthritis. Arthritis Res. Ther. 2004, 6, 107–111.
- Sokolove, J.; Brennan, M.J.; Sharpe, O.; Lahey, L.J.; Kao, A.H.; Krishnan, E.; Edmundowicz, D.; Lepus, C.M.; Wasko, M.C.; Robinson, W.H. Brief Report: Citrullination within the Atherosclerotic Plaque: A Potential Target for the Anti-Citrullinated Protein Antibody Response in Rheumatoid Arthritis. Arthritis Rheum. 2013, 65, 1719–1724.
- Geraldino-Pardilla, L.; Giles, J.T.; Sokolove, J.; Zartoshti, A.; Robinson, W.H.; Budoff, M.; Detrano, R.; Bokhari, S.; Bathon, J.M. Association of Anti-Citrullinated Peptide Antibodies with Coronary Artery Calcification in Rheumatoid Arthritis. Arthritis Rheum. 2017, 69, 1276–1281.
- Vossenaar, E.R.; Després, N.; Lapointe, E.; Van Der Heijden, A.; Lora, M.; Senshu, T.; Van Venrooij, W.J.; Ménard, H.A. Rheumatoid arthritis specific anti-Sa antibodies target citrullinated vimentin. Arthritis Res. Ther. 2004, 6, R142–R150.
- Bang, H.; Egerer, K.; Gauliard, A.; Lüthke, K.; Rudolph, P.E.; Fredenhagen, G.; Berg, W.; Feist, E.; Burmester, G.-R.; Burmester, G. Mutation and citrullination modifies vimentin to a novel autoantigen for rheumatoid arthritis. Arthritis Rheum. 2007, 56, 2503–2511.
- György, B.; Tóth, E.; Tarcsa, E.; Falus, A.; Buzás, E.I. Citrullination: A posttranslational modification in health and disease. Int. J. Biochem. Cell Biol. 2006, 38, 1662–1677.
- Van Venrooij, W.J.; Pruijn, G.J. How citrullination invaded rheumatoid arthritis research. Arthritis Res. Ther. 2014, 16, 103.
- Helin, H.J.; Korpela, M.M.; Mustonen, J.T.; Pasternack, A.I. Renal biopsy findings and clinicopathologic correlations in rheumatoid arthritis. Arthritis Rheum. 1995, 38, 242–247.
- Paudyal, S.; Yang, F.M.; Rice, C.; Chen, C.-C.; Skelton, M.; Bethel, M.; Brown, S.; Nahman, N.S.; Carbone, L. End-stage renal disease in patients with rheumatoid arthritis. Semin. Arthritis Rheum. 2017, 46, 418–422.
- Kapoor, T.; Bathon, J. Renal Manifestations of Rheumatoid Arthritis. Rheum. Dis. Clin. N. Am. 2018, 44, 571–584.
- Anders, H.-J.; Vielhauer, V. Renal co-morbidity in patients with rheumatic diseases. Arthritis Res. Ther. 2011, 13, 222.
- Zetterholm, S.G.; Verville, G.A.; Boutwell, L.; Boland, C.; Prather, J.C.; Bethea, J.; Cauley, J.; Warren, K.E.; Smith, S.A.; Magers, D.H.; et al. Noncovalent Interactions between Trimethylamine N-Oxide (TMAO), Urea, and Water. J. Phys. Chem. B 2018, 122, 8805–8811.
- Ganguly, P.; Boserman, P.; van der Vegt, N.F.A.; Shea, J.E. Trimethylamine N-Oxide Counteracts Urea Denaturation by Inhibiting Protein-Urea Preferential Interaction. J. Am. Chem. Soc. 2018, 140, 483–492.
- Shepshelovich, J.; Goldstein-Magal, L.; Globerson, A.; Yen, P.M.; Rotman-Pikielny, P.; Hirschberg, K. Protein synthesis inhibitors and the chemical chaperone TMAO reverse endoplasmic reticulum perturbation induced by overexpression of the iodide transporter pendrin. J. Cell Sci. 2005, 118, 1577–1586.
- Bchetnia, M.; Lacroix, J.; Farez, T.; Larouche, M.; Powell, J.; McCuaig, C.; Dupere, A.; Morin, C.; Legendre-Guillemin, V.; Laprise, C. Reduction in Keratin Aggregates in Epidermolysis Bullosa Simplex Keratinocytes after Pretreatment with Trimethylamine N-Oxide. Exp. Dermatol. 2016, 25, 229–230.
- Vogt, N.M.; Romano, K.A.; Darst, B.F.; Engelman, C.D.; Johnson, S.C.; Carlsson, C.M.; Asthana, S.; Blennow, K.; Zetterberg, H.; Bendlin, B.B.; et al. The gut microbiota-derived metabolite trimethylamine N-oxide is elevated in Alzheimer’s disease. Alzheimer’s Res. Ther. 2018, 10, 124.
- Wu, W.-K.; Panyod, S.; Ho, C.-T.; Kuo, C.-H.; Wu, M.-S.; Sheen, L.-Y. Dietary allicin reduces transformation of L-carnitine to TMAO through impact on gut microbiota. J. Funct. Foods 2015, 15, 408–417.
More