Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Vivi Li and Version 1 by Maria Antonietta Blasi.
No standard treatment has been established for metastatic uveal melanoma (mUM). Immunotherapy is commonly used for this disease even though UM has not been included in phase III clinical trials with checkpoint inhibitors. Unfortunately, only a minority of patients obtain a clinical benefit with immunotherapy. The immunological features of mUM were reviewed in order to understand if immunotherapy could still play a role for this disease.
- immunotherapy
- uveal melanoma
- checkpoint inhibitors
- PD-L1
- nivolumab
- pembrolizumab
- ipilimumab
- IDO
1. Introduction
Standard treatments for metastatic uveal melanoma (mUM) have not been defined yet. Several treatments have been employed with poor results [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29][1][2][3][4][5][6][7][8][9][10][11][12][13][14][15][16][17][18][19][20][21][22][23][24][25][26][27][28][29]. UM was not included in phase III clinical trials with immunotherapy for melanoma, due to the specific biological behavior and the different clinical outcome [3,4,5,6,7,8,30,31,32,33][3][4][5][6][7][8][30][31][32][33]. Nevertheless, immune checkpoint inhibitors (ICIs) are commonly used for the treatment of metastatic UM [3,4,5,6,7,8][3][4][5][6][7][8].
The available reports with ICIs in UM showed limited results in terms of efficacy, whereas they demonstrated a favorable tolerability of these agents (Table 1).
Table 1. Clinical studies with immune checkpoint inhibitors (ICIs) in uveal melanoma.
| Authors | Treatment | Type of Study | No. of Enrolled Patients | Year |
|---|---|---|---|---|
| Zimmer et al. [19] | Ipilimumab | Phase II trial. Pre-treated and naïve patients. | 53 | 2015 |
| Maio et al. [17] | Ipilimumab | Retrospective analysis. Pre-treated patients. | 82 | 2013 |
| Kelderman et al. [20] | Ipilimumab | Retrospective analysis. Pre-treated patients. | 22 | 2013 |
| Luke et al. [15] | Ipilimumab | Retrospective, multi-center analysis. Pre-treated and naïve patients. | 39 | 2013 |
| Piulats Rodriguez et al. [21] | Ipilimumab | Phase II trial. Naïve patients | 32 | 2014 |
| Danielli et al. [10] | Ipilimumab | Retrospective analysis. Pre-treated patients. | 13 | 2012 |
| Khattak et al. [13] | Ipilimumab | Retrospective analysis, single center analysis. Pre-treated patients. | 5 | 2016 |
| Deo [22] | Ipilimumab | Retrospective, single center analysis. Pre-treated patients. | 24 | 2014 |
| Shaw et al. [23] | Ipilimumab | EAP. | 18 | 2012 |
| Joshua et al. [11] | Tremelimumab | Phase II trial. Naïve patients. | 11 | 2015 |
| Algazi et al. [9] | Pembrolizumab, Nivolumab, Atezolizumab | Retrospective, multi-center analysis. Pre-treated and naïve patients. | 56 | 2016 |
| Mignard et al [16] | Pembrolizumab, Nivolumab, Ipilimumab | Retrospective, multi-center analysis. | 100 | 2018 |
| Bender et al. [27] | Pembrolizumab, Nivolumab | Retrospective, multi-center analysis. Pre-treated patients. | 15 | 2017 |
| Heppt et al. [24] | Pembrolizumab, Nivolumab, Ipilimumab | Retrospective, multi-center analysis. Pre-treated and naïve patients. | 96 | 2017 |
| Piperno-Neumann et al. [25] | Pembrolizumab, Nivolumab | Retrospective, single center analysis. Naïve patients. | 21 | 2016 |
| Karydis et al. [12] | Pembrolizumab | Retrospective analysis. Pre-treated patients. | 25 | 2016 |
| Rossi et al. [18] | Pembrolizumab | Prospective. Naïve patients. | 17 | 2019 |
| Kottschade et al. [14] | Pembrolizumab | Retrospective, single-center analysis. Pre-treated patients. | 8 | 2016 |
| Van der Kooij et al. [26] | Pembrolizumab | Prospective. Pre-treated and naïve patients. | 17 | 2017 |
| Schadendorf et al. [27] | Nivolumab | Phase II. Pre-treated patients. | 75 | 2017 |
| Jung et al. [28] | Ipilimumab | Named patient use. Pre-treated patients. | 10 | 2017 |
| Shoushtari et al. [29] | Nivolumab, Ipilimumab | Expanded access program. | 6 | 2016 |
EAP: Expand Access Program.
Increasing evidence demonstrates that uveal melanoma cells employ escape mechanisms to elude the immune system [34]. The environment of the eye is an important immune-privileged site, with many immunosuppressive mechanisms that primary UM cells retain to gain immune-protection even when they leave the eye. The immune-modulatory microenvironment of the liver, the typical site of metastasis for UM, could further protect escaped UM cells from immune surveillance [35]. Furthermore, the low mutational burden is considered another relevant characteristic of uveal melanoma, which could justify the limited activity of immunotherapy [36].
In this review, we aim to describe the immunological features of UM in order to understand if it could still be a rationale for using immunotherapy in this disease.
2. Microenvironment of the Eye
The eye is considered a “privileged immunological site” [35] due to different mechanisms of immune protection.
The first is represented by the blood–ocular barrier: the tight junctions and the lack of lymphatic vessels in the cornea and uvea limit the circulation of immune cells [37].
The other mechanism involved is represented by soluble immunosuppressive factors in the aqueous humor [38]. They are: transforming growth factor (TGF)-β, α-melanocyte-stimulating hormone (α-MSH), calcitonin gene-related peptide (CGRP), vasoactive intestinal protein (VIP), and indoleamine 2,3 dioxygenase (IDO). TGF-β inhibits the activation of macrophages, T lymphocytes, and natural killer (NK) cells and enhances the tolerance of antigen-presenting cells (APCs) [34]. TGF-β is also required for CTLA-4 up-regulation on CD8+ T cells. CTLA-4 stimulation leads to T cell inactivation and generation of T regulatory (Treg) cells [39]. α-MSH reduces neutrophil activities and stimulates Tregs. α-MSH and CGRP downregulate the production of pro-inflammatory factors. VIP inhibits NK-cells mediated cytolysis. VIP and IDO inhibit T cell activation [34].
Moreover, the cornea, iris, and retina cells express immunosuppressive ligands on their surface, such as PD-L1 and FasL. PD-L1 suppresses proliferation and induces T-lymphocyte and neutrophil apoptosis when it recognizes its ligand, PD-1, on these cells. First apoptosis signal ligand (FasL, a member of the TNF family) promotes apoptosis of activated T cells [34]. Furthermore, complement regulatory proteins (CRPs) are capable of interrupting the complement cascade with the inhibition of complement-mediated cytolysis in the eye [40].
Corneal and retinal cells express MHC-Ib molecules (such as HLA-G and HLA-E); in this way, they are able to inhibit natural killer cell cytotoxic activity, binding the inhibitory receptors, such as CD94-NKG2. The interaction between MHC-I molecules and inhibitory receptors delivers ‘‘off signals’’ to NK cells, blocking their ability to kill target cells. On the other hand, the MHC class Ia molecules are down-regulated on corneal and retinal cells, reducing the susceptibility to T-cell mediated cytolysis [34].
When antigens enter the eye, a unique mechanism of immune privilege is involved: it is termed “anterior chamber-associated immune deviation” (ACAID), an immunomodulatory phenomenon involving the eye, but also the thymus, spleen, and sympathetic nervous system [41]. In mouse models an antigen injected into the anterior chamber of the eye is captured by ocular APCs. Then, these APCs migrate to the spleen and thymus, inducing immunomodulatory cells, such as Tregs, regulatory B cells, and NK T cells, which cause immune deviation. The sympathetic nervous system promotes the maintenance of a functional ACAID [42].
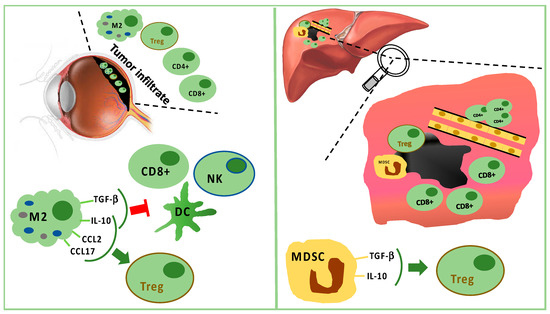 The analysis of uveal melanoma suspensions has revealed that the majority of tumor infiltrating lymphocytes (TILs) are CD8+ T cells with fewer CD4+ T cells [52][47]. Lagouros reported a limited number of Tregs in primary UM [53][48]. Nevertheless, the count of Tregs within primary tumor correlates with the development of systemic metastases. In addition, Tregs and cyclooxygenase-2 (COX-2) expression in primary tumors are associated with a poor prognosis [54][49]. Tregs are recruited by IL-10, chemokine (C–C motif) ligand (CCL) 17, and CCL22 produced by M2 macrophages [35].
Tumor-associated macrophages (TAMs) are a negative prognostic factor for UM [55][50]. Indeed, TAMs are associated with highly malignant tumors characterized by negative prognostic features, such as epithelioid cells, high microvascular density, intense pigmentation, and larger size [56][51]. TAMs seem to promote tumor growth through angiogenesis and metastatic spread [55,57][50][52].
In primary UM, the expression of HLA class I and II exerts a prognostic role [58][53]. It has been reported that, in primary tumors, down-regulation of HLA class I is a mechanism for evading CD8+ cell cytotoxicity. Therefore, tumors should be more sensitive to NK cells [59,60,61,62][54][55][56][57]. Nevertheless, only 50% of primary UM expresses MHC class I related chain (MIC) A and B, which are the ligands for NK cell receptor (NKG2D). In UM metastases, MIC A and B are absent. Consequently, the activity of NK cells in metastatic UM is limited. Vetter reported a single case of MIC expression induced after chemotherapy, suggesting a possible role for immunotherapy following cytotoxic therapy [59][54]. It has also been proved that UM cells are capable of producing the macrophage migration inhibitory factor (MIF), a cytokine that inhibits cytolytic activity of NK cells, contributing to tumor growth and metastatic spread [63,64,65][58][59][60]. The expression of FasL is a further mechanism in UM explaining the escape from NK cells [66][61]. Additionally, TILs and TAMs of UM produce IL-2 and IL-15, which bind receptors on tumor cells, promoting UM cell growth and reducing sensitivity to NK cell activity [67][62].
Moreover, the loss of the maturation marker CD-40 on APCs has been observed in primary UM. This lack does not allow correct lymphocyte T-mediated anti-tumor activity because of an inadequate functioning of APCs to induce T cell activation [68][63].
Overall, “inflammatory phenotype” has been proposed to define UM with infiltrating macrophages and lymphocytes in addition to a high expression of HLA class I and II molecules. It identifies tumors with a worse prognosis [48,49][43][44].
It is doubtless that the immune cells in the UM inflammatory phenotype do not stimulate an antitumor response. They contribute to angiogenesis, immunosuppression, tumor growth, and metastatic spread [69][64]. Moreover, comparing the circulating immune cells between primary and metastatic UM, a weaker immune surveillance has been spotted in case of metastases. Lower circulating CD3−CD56dim NK cells, CD8+, and NK T cells, as well as an increase in Tregs and MSDCs have been detected during the metastatic phase. Furthermore, metastases are associated with increasing plasma levels of several miRNAs involved in immune regulation [70][65].
In the metastatic sites, CD4+ cells are present in perivascular aggregates, while CD8+ cells are scarce and mainly surround the liver metastases [71,72,73,74][66][67][68][69]. Tumor cells and resident hepatic cells recruit in metastatic sites two different types of myeloid-derived suppressor cells (MDSCs): monocytic MDSCs and polymorphonuclear MDSCs. Monocytic MDSCs, more frequent than polymorphonuclear MDSCs in metastases, promote neoplastic growth after their differentiation towards TAMs. MDSCs are able to induce Tregs releasing IL-10 and TGF-β [35,71][35][66] (Figure 1).
Nevertheless, Rothermel found that a subset of TILs associated with a lack of melanin pigmentation is more effective in antitumor response, similarly to TILs in cutaneous melanoma [75][70].
The analysis of uveal melanoma suspensions has revealed that the majority of tumor infiltrating lymphocytes (TILs) are CD8+ T cells with fewer CD4+ T cells [52][47]. Lagouros reported a limited number of Tregs in primary UM [53][48]. Nevertheless, the count of Tregs within primary tumor correlates with the development of systemic metastases. In addition, Tregs and cyclooxygenase-2 (COX-2) expression in primary tumors are associated with a poor prognosis [54][49]. Tregs are recruited by IL-10, chemokine (C–C motif) ligand (CCL) 17, and CCL22 produced by M2 macrophages [35].
Tumor-associated macrophages (TAMs) are a negative prognostic factor for UM [55][50]. Indeed, TAMs are associated with highly malignant tumors characterized by negative prognostic features, such as epithelioid cells, high microvascular density, intense pigmentation, and larger size [56][51]. TAMs seem to promote tumor growth through angiogenesis and metastatic spread [55,57][50][52].
In primary UM, the expression of HLA class I and II exerts a prognostic role [58][53]. It has been reported that, in primary tumors, down-regulation of HLA class I is a mechanism for evading CD8+ cell cytotoxicity. Therefore, tumors should be more sensitive to NK cells [59,60,61,62][54][55][56][57]. Nevertheless, only 50% of primary UM expresses MHC class I related chain (MIC) A and B, which are the ligands for NK cell receptor (NKG2D). In UM metastases, MIC A and B are absent. Consequently, the activity of NK cells in metastatic UM is limited. Vetter reported a single case of MIC expression induced after chemotherapy, suggesting a possible role for immunotherapy following cytotoxic therapy [59][54]. It has also been proved that UM cells are capable of producing the macrophage migration inhibitory factor (MIF), a cytokine that inhibits cytolytic activity of NK cells, contributing to tumor growth and metastatic spread [63,64,65][58][59][60]. The expression of FasL is a further mechanism in UM explaining the escape from NK cells [66][61]. Additionally, TILs and TAMs of UM produce IL-2 and IL-15, which bind receptors on tumor cells, promoting UM cell growth and reducing sensitivity to NK cell activity [67][62].
Moreover, the loss of the maturation marker CD-40 on APCs has been observed in primary UM. This lack does not allow correct lymphocyte T-mediated anti-tumor activity because of an inadequate functioning of APCs to induce T cell activation [68][63].
Overall, “inflammatory phenotype” has been proposed to define UM with infiltrating macrophages and lymphocytes in addition to a high expression of HLA class I and II molecules. It identifies tumors with a worse prognosis [48,49][43][44].
It is doubtless that the immune cells in the UM inflammatory phenotype do not stimulate an antitumor response. They contribute to angiogenesis, immunosuppression, tumor growth, and metastatic spread [69][64]. Moreover, comparing the circulating immune cells between primary and metastatic UM, a weaker immune surveillance has been spotted in case of metastases. Lower circulating CD3−CD56dim NK cells, CD8+, and NK T cells, as well as an increase in Tregs and MSDCs have been detected during the metastatic phase. Furthermore, metastases are associated with increasing plasma levels of several miRNAs involved in immune regulation [70][65].
In the metastatic sites, CD4+ cells are present in perivascular aggregates, while CD8+ cells are scarce and mainly surround the liver metastases [71,72,73,74][66][67][68][69]. Tumor cells and resident hepatic cells recruit in metastatic sites two different types of myeloid-derived suppressor cells (MDSCs): monocytic MDSCs and polymorphonuclear MDSCs. Monocytic MDSCs, more frequent than polymorphonuclear MDSCs in metastases, promote neoplastic growth after their differentiation towards TAMs. MDSCs are able to induce Tregs releasing IL-10 and TGF-β [35,71][35][66] (Figure 1).
Nevertheless, Rothermel found that a subset of TILs associated with a lack of melanin pigmentation is more effective in antitumor response, similarly to TILs in cutaneous melanoma [75][70].
3. Immune Infiltrate in Uveal Melanoma
The crosstalk between tumor and microenvironment influences the inflammatory response: cancer cells interact with both the innate and the adaptive immune system and use immune cells for tumor survival and protection from immunological attacks. The main immune cells in uveal melanoma are the M2-type macrophages, which foster tumor growth through angiogenesis and immunosuppression [48,49][43][44]. Monocytes/macrophages of M2 lineage exceed T cells in tumor-infiltrating immune cells. They secrete anti-inflammatory cytokines (IL-10 and TGF-β), which inhibit dendritic cell activation, as well as T- and NK-cell functions [35,48][35][43] (Figure 1). Treg and UM cells trigger inflammation and M2-polarization through the production of factors, such as CCL22, CCL2, VEGF, M-CSF, TGF-β, IL-6, IL-10, CCL17, CCL22, PGE2, and endothelial monocyte-activating polypeptide (EMAP)-II [35,48,50][35][43][45]. Moreover, M2-macrophages can secrete soluble factors, which enhance the invasive capabilities of neoplastic cells, such as the melanoma inhibitory activity (MIA), a growth-regulatory protein, which inhibits the cellular adhesion to the extracellular matrix [51][46].Figure 1. Tumor infiltrating immune cells in primary tumor (left panel) and in liver metastasis (right panel). In primary uveal melanoma (UM), CD8+ T cells and fewer CD4+ T cells are present, but M2 polarized macrophages are predominant. M2 macrophages stimulate Tregs through IL-10, CCL2, and CCL17. M2 macrophages suppress CD8+, NK, and DC secreting TGF-beta and IL-10. In liver metastasis, CD8+ cells surround but do not infiltrate the tumor mass, while CD4+ cells are present in perivascular aggregates. Furthermore, Tregs are stimulated by TGF-beta and IL-10 produced by MDSC.
3.1. The Role of PD-1/PD-L1 Interaction in UM
UM cell lines are able to suppress T-cell activation by the expression of PD-L1 (B7-H1) [76,77][71][72]. However, in vivo a limited constitutive expression of PD-L1 on tumor cells and PD-1 on TILs has been proven in metastatic uveal melanoma compared with cutaneous melanoma [78][73]. As a matter of fact, in the metastatic sites, only 5% of uveal melanoma shows the expression of PD-L1, whereas PD-1 is expressed in about 51% of TILs [79][74]. Regarding primary UM, 40% of PD-L1 expression on tumor cells is reported in patients with metastatic disease [80][75].
PD-L1 expression indicates an active interaction between the tumor and the adaptive immune cells. PD-L1 expression is induced by IFN-γ produced by activated CD8+ cells. Consequently, PD-L1 on tumor cells depends on the presence of activated TILs PD-1+ [79][74]. The lower PD-L1 expression in UM is not due to a loss of function. Indeed, it is known that uveal melanoma cells do not lose the ability to upregulate PD-L1 in response to IFN-γ. In a preclinical model, PD-L1 is able to inhibit T cell proliferation through a lower secretion of IL2 [81][76].
Among PD-1/PD-L1 patterns, in UM samples the two more frequent patterns are PD-1−/PD-L1− and PD-1+/PD-L1−, representing immunological tolerance, with, respectively, absence or functional suppression of TILs in the tumor microenvironment. Differently, in cutaneous melanoma the dominant subgroup is the PD-1+/PD-L1+, resulting in immune-competent and active TILs. This subgroup seems to be absent in UM. TILs with antitumor function are less frequent than regulatory TILs in UM, resulting in immune tolerance both in the primary site and metastases. [79][74]. PD-1/PD-L1 axis is probably not one of the most relevant mechanisms to avoid immune response in uveal melanoma. This hypothesis can explain the poor response to anti-PD-1 therapy.
3.2. IDO and Immune Escape
The enzyme indoleamine 2,3 dioxygenase (IDO) controls tryptophan degradation, influencing both the innate and the adaptive immune system for lymphocytes proliferation, activation, and survival. IDO is able to suppress T and NK cells, generate Tregs, and promote tumor angiogenesis.
In uveal melanoma IDO is not constitutively expressed either in primary or in metastatic cells. However, IFN-γ upregulates IDO mRNA and protein in UM cells, inducing the production of enzymatically and biologically active IDO [82,83][77][78]. We might conclude that the induction of IDO by IFN-γ is a defensive mechanism of uveal melanoma cells in response to the presence of T lymphocytes and NK cells. Upregulation of the IDO gene and protein expression can contribute to an immune-privileged microenvironment promoting immune escape. In advanced cutaneous melanoma, epacadostat, an IDO1 inhibitor, in addition to anti-PD-1 treatment, does not improve the clinical outcomes of anti-PD-1 therapy alone [84][79]. The effectiveness of IDO inhibitors in UM could be limited by the lack of a constitutive expression. Nevertheless, we cannot exclude a role of IDO inhibitors in selected patients or in special conditions inducing IDO expression (i.e., previous treatments).
References
- Carvajal, R.D.; Piperno-Neumann, S.; Kapiteijn, E.; Chapman, P.B.; Frank, S.; Joshua, A.M.; Piulats, J.M.; Wolter, P.; Cocquyt, V.; Chmielowski, B.; et al. Selumetinib in combination with dacarbazine in patients with metastatic uveal melanoma: A phase III, multicenter, randomized trial (SUMIT). J. Clin. Oncol. 2018, 36, 1232–1239.
- Schinzari, G.; Rossi, E.; Cassano, A.; Dadduzio, V.; Quirino, M.; Pagliara, M.; Blasi, M.A.; Barone, C. Cisplatin, dacarbazine and vinblastine as first line chemotherapy for liver metastatic uveal melanoma in the era of immunotherapy: A single institution phase II study. Melanoma Res. 2017, 27, 591–595.
- Buder, K.; Gesierich, A.; Gelbrich, G.; Goebeler, M. Systemic treatment of metastatic uveal melanoma: Review of literature and future perspectives. Cancer Med. 2013, 2, 674–686.
- Komatsubara, K.M.; Carvajal, R.D. Immunotherapy for the treatment of uveal melanoma: Current status and emerging therapies. Curr. Oncol. Rep. 2017, 19, 45.
- Oliva, M.; Rullan, A.J.; Piulats, J.M. Uveal melanoma as a target for immune-therapy. Ann. Transl. Med. 2016, 4, 172.
- Singh, A.D.; Turell, M.E.; Topham, A.K. Uveal melanoma: Trends in incidence, treatment, and survival. Ophthalmology 2011, 118, 1881–1885.
- Spagnolo, F.; Picasso, V.; Spano, L.; Tanda, E.; Venzano, C.; Queirolo, P. Update on metastatic uveal melanoma: Progress and challenges. BioDrugs 2016, 30, 161–172.
- Bender, C.; Enk, A.; Gutzmer, R.; Hasser, C. Anti-PD-1 antibodies in metastatic uveal melanoma: A treatment option? Cancer Med. 2017, 6, 1581–1586.
- Algazi, A.P.; Tsai, K.K.; Shoushtari, A.N.; Munhoz, R.R.; Eroglu, Z.; Piulats, J.M.; Ott, P.A.; Johnson, D.B.; Hwang, J.; Daud, A.I.; et al. Clinical outcomes in metastatic uveal melanoma treated with PD-1 and PD-L1 antibodies. Cancer 2016, 122, 3344–3353.
- Danielli, R.; Ridolfi, R.; Chiarion-Sileni, V.; Queirolo, P.; Testori, A.; Plummer, R.; Boitano, M.; Calabrò, L.; Rossi, C.D.; Giacomo, A.M.; et al. Ipilimumab in pretreated patients with metastatic uveal melanoma: Safety and clinical efficacy. Cancer Immunol. Immunother. 2012, 61, 41–48.
- Joshua, A.M.; Monzon, J.G.; Mihalcioiu, C.; Hogg, D.; Smylie, M.; Cheng, T. A phase 2 study of tremelimumab in patients with advanced uveal melanoma. Melanoma Res. 2015, 25, 342–347.
- Karydis, I.; Chan, P.Y.; Wheater, M.; Arriola, E.; Szlosarek, P.W.; Ottensmeier, C.H. Clinical activity and safety of pembrolizumab in ipilimumab pre-treated patients with uveal melanoma. Oncoimmunology 2016, 5, e1143997.
- Khattak, M.A.; Fisher, R.; Hughes, P.; Gore, M.; Larkin, J. Ipilimumab activity in advanced uveal melanoma. Melanoma Res. 2013, 23, 79–81.
- Kottschade, L.A.; McWilliams, R.R.; Markovic, S.N.; Block, M.S.; Villasboas Bisneto, J.; Pham, A.Q.; Esplin, B.L.; Dronca, R.S. The use of pembrolizumab for the treatment of metastatic uveal melanoma. Melanoma Res. 2016, 26, 300–303.
- Luke, J.J.; Callahan, M.K.; Postow, M.A.; Romano, E.; Ramaiya, N.; Bluth, M.; Giobbie-Hurder, A.; Lawrence, D.P.; Ibrahim, N.; Ott, P.A.; et al. Clinical activity of ipilimumab for metastatic uveal melanoma: A retrospective review of the Dana-Farber Cancer Institute, Massachusetts General Hospital, Memorial Sloan-Kettering Cancer Center, and University Hospital of Lausanne experience. Cancer 2013, 119, 3687–3695.
- Mignard, C.; Deschamps Huvier, A.; Gillibert, A.; Duval Modeste, A.B.; Dutriaux, C.; Khammari, A.; Avril, M.F.; Kramkimel, N.; Mortier, L.; Marcant, P.; et al. Efficacy of Immunotherapy in Patients with Metastatic Mucosal or Uveal Melanoma. J. Oncol. 2018, 2018, 1908065.
- Maio, M.; Danielli, R.; Chiarion-Sileni, V.; Pigozzo, J.; Parmiani, G.; Ridolfi, R.; De Rosa, F.; Del Vecchio, M.; Di Guardo, L.; Queirolo, P.; et al. Efficacy and safety of ipilimumab in patients with pre-treated, uveal melanoma. Ann. Oncol. 2013, 24, 2911–2915.
- Rossi, E.; Pagliara, M.M.; Orteschi, D.; Dosa, T.; Sammarco, M.G.; Caputo, C.G.; Petrone, G.; Rindi, G.; Zollino, M.; Blasi, M.A.; et al. Pembrolizumab as first-line treatment for metastatic uveal melanoma. Cancer Immunol. Immunother. 2019, 68, 1179–1185.
- Zimmer, L.; Vaubel, J.; Mohr, P.; Hauschild, A.; Utikal, J.; Simon, J. Phase II DeCOG-study of ipilimumab in pretreated and treatment naive patients with metastatic uveal melanoma. PLoS ONE 2015, 10, e0118564.
- Kelderman, S.; van der Kooij, M.K.; van den Eertwegh, A.J.; Soetekouw, P.M.; Jansen, R.L.; van den Brom, R.R.; Hospers, G.A.; Haanen, J.B.; Kapiteijn, E.; Blank, C.U. Ipilimumab in pretreated metastastic uveal melanoma patients. Results of the Dutch Working group on Immunotherapy of Oncology (WIN-O). Acta Oncol. 2013, 52, 1786–1788.
- Piulats Rodriguez, J.M.; Ochoa de Olza, M.; Codes, M.; Lopez-Martin, J.A.; Berrocal, A.; García, M.; Gurpide, A.; Homet, B.; Martin-Algarra, S. Phase II study evaluating ipilimumab as a single agent in the first-line treatment of adult patients (Pts) with metastatic uveal melanoma (MUM): The GEM-1 trial. J. Clin. Oncol. 2014, 32, 9033.
- Deo, M.A. Long-term survival benefit from ipilimumab treatment in metastatic uveal melanoma patients. J. Clin. Oncol. 2014, 32, 3060.
- Shaw, H.; Larkin, J.; Corrie, P. Ipilimumab for advanced melanoma in an expanded access program (EAP): Ocular, mucosal and acral subtype UK experience. Ann. Oncol. 2012, 23, 374.
- Heppt, M.V.; Heinzerling, L.; Kähler, K.C.; Forschner, A.; Kirchberger, M.C.; Loquai, C.; Meissner, M.; Meier, F.; Terheyden, P.; Schell, B.; et al. Prognostic factors and outcomes in metastatic uveal melanoma treated with programmed cell death-1 or combined PD-1/cytotoxic T-lymphocyte antigen-4 inhibition. Eur. J. Cancer 2017, 82, 56–65.
- Piperno-Neumann, S.; Servois, V.; Mariani, P.; Cassoux, N.; Barnhill, R.; Rodrigues, M.J. Activity of anti-PD1 drugs in uveal melanoma patients. J. Clin. Oncol. 2016, 34, 9588.
- Van der Kooij, M.K.; Joosse, A.; Speetjens, F.M.; Hospers, G.A.; Bisschop, C.; de Groot, J.W.; Koornstra, R.; Blank, C.U.; Kapiteijn, E. Anti-PD1 treatment in metastatic uveal melanoma in the Netherlands. Acta Oncol. 2017, 56, 101–103.
- Schadendorf, D.; Ascierto, P.A.; Haanen, J.; Espinosa, E.; Demidov, L.V.; Garbe, C.; Lorigan, P.; Gogas, H.; Hoeller, C.; Guren, T.K.; et al. Efficacy and safety of nivolumab (NIVO) in patients with advanced melanoma (MEL) and poor prognostic factors who progressed on or after ipilimumab (IPI): Results from a phase II study (CheckMate 172). J. Clin. Oncol. 2017, 35, 9524.
- Jung, M.; Lee, J.; Kim, T.M.; Lee, D.H.; Kang, J.H.; Oh, S.Y.; Lee, S.J.; Shin, S.J. Ipilimumab real-world efficacy and safety in korean melanoma patients from the korean named patient program cohort. Cancer Res. Treat. 2017, 49, 44–53.
- Shoushtari, A.N.; Navld-Azarbaljanl, P.; Friedman, C.F.; Panageas, K.; Postow, M.A.; Callahan, M.K.; Momtaz, P.; Campbell, S.C.; Shames, Y.; Prempeh-Keteku, N.A.; et al. Efficacy of nivolumab and ipilimumab (Nivo + Ipi) combination in melanoma patients (pts) treated at a single institution on an expanded-access program (EAP). J. Clin. Oncol. 2016, 34, 9554.
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330.
- Robert, C.; Ribas, A.; Wolchok, J.D.; Hodi, F.S.; Hamid, O.; Kefford, R. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: A randomised dose-comparison cohort of a phase 1 trial. Lancet 2014, 384, 1109–1117.
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532.
- Weber, J.S.; D’Angelo, S.P.; Minor, D.; Hodi, F.S.; Gutzmer, R.; Neyns, B.; Hoeller, C.; Khushalani, N.I.; Miller, W.H., Jr.; Lao, C.D.; et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015, 16, 375–384.
- Niederkorn, J.Y. Immune escape mechanisms of intraocular tumors. Progr. Retin. Eye Res. 2009, 28, 329–347.
- Terai, M.; Mastrangelo, M.J.; Sato, T. Immunological aspect of the liver and metastatic uveal melanoma. J. Cancer Metastasis Treat. 2017, 3, 231–243.
- Furney, S.J.; Pedersen, M.; Gentien, D.; Dumont, A.G.; Rapinat, A.; Desjardins, L.; Turajlic, S.; Piperno-Neumann, S.; de la Grange, P.; Roman-Roman, S.; et al. SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov. 2013, 3, 1122–1129.
- Bill, A. Ocular circulation. In Adler’s Physiology of the Eye, 5th ed.; Moses, R.A., Ed.; Mosby Co: St. Louis, MO, USA, 1970; pp. 278–296.
- Taylor, A.W. Ocular immunosuppressive microenvironment. Chem. Immunol. 2008, 92, 71–85.
- Sugita, S.; Ng, T.F.; Lucas, P.J.; Gress, R.E.; Streilein, J.W. B7+ iris pigment epithelium induce CD8+ T regulatory cells; both suppress CTLA-4+T cells. J. Immunol. 2006, 176, 118–127.
- Sohn, J.H.; Kaplan, H.J. Chronic low level complement activation within the eye is controlled by intraocular complement regulatory proteins. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3492–3502.
- Stein-Streilein, J.; Streilein, J.W. Anterior chamber associated immune deviation (ACAID): Regulation, biological relevance, and implications for therapy. Int. Rev. Immunol. 2002, 21, 123–152.
- Vendomèle, J.; Khebizi, Q.; Fisson, S. Cellular and molecular mechanisms of Anterior Chamber-Associated Immune Deviation (ACAID): What we have learned from knockout mice. Front. Immunol. 2017, 8, 1686.
- Bronkhorst, I.H.; Jager, M.J. Uveal melanoma: The inflammatory microenvironment. J. Innate Immun. 2012, 4, 454–462.
- Jager, M.J.; Ly, L.V.; El Filali, M.; Madigan, M.C. Macrophages in uveal melanoma and in experimental ocular tumor models: Friends or foes? Prog. Retin. Eye Res. 2011, 30, 129–146.
- Clarijs, R.; Schalkwijk, L.; Ruiter, D.J.; de Waal, R.M. EMAP-II expression is associated with macrophage accumulation in primary uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2003, 44, 1801–1806.
- Callejo, S.A.; Marshall, J.C.; Cools-Lartigue, J.; Saraiva, V.S.; Burnier, M.N., Jr. Macrophage-derived soluble factor enhances melanoma inhibitory activity expression by uveal melanoma cells In Vitro. Melanoma Res. 2004, 14, 91–95.
- Meecham, W.J.; Char, D.H.; Kaleta-Michaels, S. Infiltrating lymphocytes and antigen expression in uveal melanoma. Ophthalmic Res. 1992, 24, 20–26.
- Lagouros, E.; Salomao, D.; Thorland, E.; Hodge, D.O.; Vile, R.; Pulido, J.S. Infiltrative T regulatory cells in enucleated uveal melanomas. Trans. Am. Ophthalmol. Soc. 2009, 107, 223–228.
- Mougiakakos, D.; Johansson, C.C.; Trocme, E.; All-Ericsson, C.; Economou, M.A.; Larsson, O.; Seregard, S.; Kiessling, R. Intratumoral forkhead box P3-positive regulatory T cells predict poor survival in cyclooxygenase-2-positive uveal melanoma. Cancer 2010, 116, 2224–2233.
- Bronkhorst, I.H.; Ly, L.V.; Jordanova, E.S.; Vrolijk, J.; Versluis, M.; Luyten, G.P.; Jager, M.J. Detection of M2-macrophages in uveal melanoma and relation with survival. Investig. Ophthalmol. Vis. Sci. 2011, 52, 643–650.
- Makitie, T.; Summanen, P.; Tarkkanen, A.; Kivela, T. Tumor-infiltrating macrophages (CD68(+) cells) and prognosis in malignant uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1414–1421.
- Ly, L.V.; Baghat, A.; Versluis, M.; Jordanova, E.S.; Luyten, G.P.M.; van Rooijen, N.; van Hall, T.; van der Velden, P.A.; Jager, M.J. In aged mice, outgrowth of intraocular melanoma depends on proangiogenic M2-type macrophages. J. Immunol. 2010, 185, 3481–3488.
- Ericsson, C.; Seregard, S.; Bartolazzi, A.; Levitskaya, E.; Ferrone, S.; Kiessling, R.; Larssonet, O. Association of HLA class I and class II antigen expression and mortality in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2153–2156.
- Vetter, C.S.; Lieb, W.; Brocker, E.B.; Becker, J.C. Loss of nonclassical MHC molecules MIC-A/B expression during progression of uveal melanoma. Br. J. Cancer 2004, 91, 1495–1499.
- Jager, M.J.; Hurks, H.M.; Levitskaya, J.; Kiessling, R. HLA expression in uveal melanoma: There is no rule without some exception. Hum. Immunol. 2002, 63, 444–451.
- Blom, D.J.; Luyten, G.P.; Mooy, C.; Kerkvliet, S.; Zwinderman, A.H.; Jager, M.J. Human leukocyte antigen class I expression. Marker of poor prognosis in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 1997, 38, 1865–1872.
- De Waard-Siebinga, I.; Hilders, C.G.; Hansen, B.E.; van Delft, J.L.; Jager, M.J. HLA expression and tumor-infiltrating immune cells in uveal melanoma. Graefes Arch. Clin. Exp. Ophthalmol. 1996, 234, 34–42.
- Repp, A.C.; Mayhew, E.S.; Apte, S.; Niederkorn, J.Y. Human uveal melanoma cells produce macrophage migration-inhibitory factor to prevent lysis by NK cells. J. Immunol. 2000, 165, 710–715.
- Apte, R.S.; Mayhew, E.; Niederkorn, J.Y. Local inhibition of natural killer cell activity promotes the progressive growth of intraocular tumors. Investig. Ophthalmol. Vis. Sci. 1997, 38, 1277–1282.
- Apte, R.S.; Sinha, D.; Mayhew, E.; Wistow, G.J.; Niederkorn, J.Y. Cutting edge: Role of macrophage migration inhibitory factor in inhibiting NK cell activity and preserving immune privilege. J. Immunol. 1998, 160, 5693–5696.
- Anastassiou, G.; Coupland, S.E.; Stang, A.; Boeloeni, R.; Schilling, H.; Bornfeld, N. Expression of Fas and Fas ligand in uveal melanoma: Biological implication and prognostic value. J. Pathol. 2001, 194, 466–472.
- He, Y.G.; Mayhew, E.; Mellon, J.; Niederkorn, J.Y. Expression and possible function of IL-2 and IL-15 receptors on human uveal melanoma cells. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4240–4246.
- Polak, M.E.; Borthwick, N.J.; Johnson, P.; Hungerford, J.L.; Higgins, B.; Di Palma, S.; Jager, M.J.; Cree, I.A. Presence and phenotype of dendritic cells in uveal melanoma. Br. J. Ophthalmol. 2007, 91, 971–976.
- Bronkhorst, I.H.; Vu, T.H.; Jordanova, E.S.; Luyten, G.P.; Burg, S.H.; Jager, M.J. Different subsets of tumor-infiltrating lymphocytes correlate with macrophage influx and monosomy 3 in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2012, 53, 5370–5378.
- Achberger, S.; Aldrich, W.; Tubbs, R.; Crabb, J.W.; Singh, A.D.; Triozzi, P.L. Circulating immune cell and microRNA in patients with uveal melanoma developing metastatic disease. Mol. Immunol. 2014, 58, 182–186.
- Krishna, Y.; McCarthy, C.; Kalirai, H.; Coupland, S.E. Inflammatory cell infiltrates in advanced metastatic uveal melanoma. Hum. Pathol. 2017, 66, 159–166.
- Whelchel, J.C.; Farah, S.E.; McLean, I.W.; Burnier, M.N. Immunohistochemistry of infiltrating lymphocytes in uveal malignant melanoma. Investig. Ophthalmol. Vis. Sci. 1993, 34, 2603–2606.
- De la Cruz, P.O., Jr.; Specht, C.S.; McLean, I.W. Lymphocytic infiltration in uveal malignant melanoma. Cancer 1990, 65, 112–115.
- Durie, F.H.; Campbell, A.M.; Lee, W.R.; Damato, B.E. Analysis of lymphocytic infiltration in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 1990, 31, 2106–2110.
- Rothermel, L.D.; Sabesan, A.C.; Stephens, D.J.; Chandran, S.S.; Paria, B.C.; Srivastava, A.K.; Somerville, R.; Wunderlich, J.R.; Lee, C.C.; Xi, L.; et al. Identification of an immunogenic subset of metastatic uveal melanoma. Clin. Cancer Res. 2016, 22, 2237–2249.
- Jia, R.; Jiao, Z.; Xu, X. Functional significance of B7-H1 expressed by human uveal melanoma cells. Mol. Med. Rep. 2011, 4, 163–167.
- Ma, J.; Usui, Y.; Kezuka, T.; Okunuki, Y.; Zhang, L.; An, X.; Mizota, A.; Goto, H. Costimulatory molecule expression on human uveal melanoma cells: Functional analysis of CD40 and B7-H1. Exp. Eye Res. 2012, 96, 98–110.
- Qin, Y.; Petaccia de Macedo, M.; Reuben, A.; Forgeta, M.A.; Haymaker, C.; Bernatcheza, C.; Spencerd, C.N.; Gopalakrishnan, V.; Reddy, S.; Cooper, Z.A.; et al. Parallel profiling of immune infiltrate subsets in uveal melanoma versus cutaneous melanoma unveils similarities and differences: A pilot study. Oncoimmunology 2017, 6, e1321187.
- Javed, A.; Arguello, D.; Johnston, C.; Gatalica, Z.; Terai, M.; Weight, R.M.; Orloff, M.; Mastrangelo, M.J.; Sato, T. PD-L1 expression in tumor metastasis is different between uveal melanoma and cutaneous melanoma. Immunotherapy 2017, 9, 1323–1330.
- Zoroquiain, P.; Esposito, E.; Logan, P.; Aldrees, S.; Dias, A.B.; Mansure, J.J.; Santapau, D.; Garcia, C.; Saornil, M.A.; Belfort Neto, R.; et al. Programmed cell death ligand-1 expression in tumor and immune cells is associated with better patient outcome and decreased tumor-infiltrating lymphocytes in uveal melanoma. Mod. Pathol. 2018, 31, 1201–1210.
- Yang, W.; Chen, P.W.; Li, H.; Alizadeh, H.; Niederkorn, J.Y. PD-L1: PD-1 interaction contributes to the functional suppression of T-Cell responses to human uveal melanoma cells In Vitro. Investig. Ophtalmol. Vis. Sci. 2008, 49, 2518–2525.
- Chen, P.; Mellon, J.K.; Mayhew, E.; Wang, S.; He, Y.G.; Hogan, N.; Niederkorn, J.Y. Uveal melanoma expression of indoleamine 2,3-deoxygenase: Establishment of an immune privileged environment by tryptophan depletion. Exp. Eye Res. 2007, 85, 617–625.
- Ryu, Y.H.; Kim, J.C. Expression of indoleamine 2,3-dioxygenase in human corneal cells as a local immunosuppressive factor. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4148–4152.
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019.
More