Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Vivi Li and Version 1 by Julia Leschik.
Newborn neurons in the adult hippocampus are regulated by many intrinsic and extrinsic cues. It is well accepted that elevated glucocorticoid levels lead to downregulation of adult neurogenesis, which this review discusses as one reason why psychiatric diseases, such as major depression, develop after long-term stress exposure. In reverse, adult neurogenesis has been suggested to protect against stress-induced major depression, and hence, could serve as a resilience mechanism.
- adult neurogenesis
- stress
- major depressive disorder
- resilience
1. Introduction: Adult Neurogenesis
Adult neurogenesis in the mammalian brain is a continuous lifelong physiological process, which dramatically declines during aging [1]. The main work, so far, elucidating the regulatory mechanisms of adult neural stem cells has been done in rodent animal models, whereas the existence of neurogenesis in the adult human brain is still under debate [2,3,4][2][3][4]. Even if many studies report the existence of adult neurogenesis in humans during the whole lifespan [5,6,7,8,9[5][6][7][8][9][10],10], these findings have been questioned by others, which could detect adult neural stem cells and their progeny only in early childhood [11,12,13][11][12][13]. The discrepancy here might arise from different probe sampling and further technical issues, which are extensively described in recent reviews by Lucassen and colleagues [14,15][14][15]. Especially, a direct comparison to rodent models seems difficult as brains from healthy human subjects cannot be processed and analyzed similarly to rodents, since human samples usually arise from postmortem fixed tissue [16].
1.1. Adult Hippocampal Neurogenesis
The generation of newly built neurons needs to be tightly controlled under physiological conditions. Control mainly occurs on three different levels, which comprise first the proliferation of adult neural stem cells and/or progenitor cells (NPCs), also maintaining the stem cell pool; second, the neuronal and glial determination and differentiation of NPCs. Lastly, newly built neurons need to survive, mature, and functionally integrate into already existing neuronal circuits, which is the final level of regulation. For a detailed description of adult neural stem cell regulation, see the two recent reviews from Obernier and Alvarez-Buylla (2019) and Denoth-Lippuner and Jessberger (2021) [17,18][17][18]. In the adult mammalian brain, two main regions are described where new neurons are continuously generated under physiological conditions. This is, on one hand, the subventricular zone (SVZ) of the lateral ventricles, which gives rise to new GABAergic granular and periglomerular neurons of the olfactory bulb. The second main neurogenic region is located in the adult hippocampus, specifically in the subgranular zone (SGZ) of the dentate gyrus (DG), which serves as an input station into the whole hippocampal formation. After cell division in the SGZ, hippocampal neural stem cells differentiate into postmitotic glutamatergic cells of the DG granule cell layer, a process which takes approximately two months from cell birth until the end of maturation [19].
The vast majority of newly built granule neurons are added to the granule cell layer of the DG throughout life, thereby extending the granule cell layer [20]. This implicates a high level of neuronal plasticity, as the addition of new neurons has possibly the capacity to rewire an existing neuronal circuit.
1.2. Adult Hippocampal Neurogenesis in Stress-Related Behavior
Given that the DG is part of the hippocampus, hippocampal function is modulated by changes in rates of adult neurogenesis. Indeed, in most studies, an increase of adult neurogenesis led to enhanced performance in hippocampal-dependent behavioral tasks, whereas a lack or reduction induced impaired hippocampal-dependent tasks [20,21,22,23,24,25][20][21][22][23][24][25]. The hippocampus is part of the limbic system and can be subdivided into the dorsal and the ventral part, both of which exert differential functions. Whereas, the ventral hippocampus is mainly important for mood control and regulating emotional states, the dorsal hippocampus has been predominantly implicated in cognitive functions, such as learning and memory [26,27,28][26][27][28]. Nonetheless, recent studies tend to show that rather than strictly containing dissociated roles, both the dorsal and the ventral hippocampus contribute to the integration of contextual information and context-specific events in a complementary way [29,30][29][30]. The hippocampus is regarded as the key brain area involved in regulating stress response [31]. Therefore, proper stress coping is associated with hippocampal processing of emotional and cognitive information. Appropriate stress coping of an individual is important to adopt or to stay in a resilient state as a protective mechanism against symptoms of stress-related psychiatric disorders, such as major depressive disorder (MDD), anxiety disorders, as well as posttraumatic stress disorder (PTSD) [32]. Particularly, adult-born DG granule cells are essential for hippocampal-dependent tasks involving pattern separation, cognitive flexibility, and memory interference, as well as forgetting [33,34,35,36][33][34][35][36]. All these processes may be relevant for the acquisition of stress resilient outcomes, and their failure could result in stress-related mental dysfunctions. Experiments ablating or reducing adult neurogenesis have demonstrated, besides a lack of spatial memory, the occurrence of depression- and anxiety-like behavior, which, however, in several studies is only detectable in response to stress [20,25,36,37,38,39,40][20][25][36][37][38][39][40]. Certainly, increasing neurogenesis is sufficient to reduce anxiety and depression-like behaviors [41] and hypothalamus pituitary adrenal (HPA) axis dysregulation [42]. In addition, the finding that rodents showing depressive-like behavior and depressed human individuals display a thinner granule cell layer, whereby antidepressant-treatment restore adult neurogenesis to physiological levels [36[36][43],43], suggests adult neurogenesis as a resilience mechanism [44]. In fact, Anacker et al. (2018) [45] recently demonstrated that young adult-born DG granule cells are necessary to confer stress resilience by inhibiting ventral mature granule neurons during chronic social defeat stress (CSDS). In line with this, a direct causal relationship between newborn neuronal activity and affective behavior was demonstrated by Tunc-Ozcan et al. (2019) [46]. The authors reported that activating newborn neurons alleviated depressive-like behavior and reversed the effects of chronic unpredictable stress (CUS). The results further suggest that the mere numbers of newborn neurons are a relatively coarse read-out, but also their neuronal activity and degree of functional integration into the existing neuronal network of the mature DG is a crucial factor in governing resilience. Modulation of network activity particularly applies to young adult-born DG granule cells in the age of 4–6 weeks after cell birth. At four weeks, young newborn DG neurons start to enter a critical period of development with distinct electrophysiological properties, including high input resistance and a lack of GABAergic inhibition, which results in a greater propensity for hyperexcitability and a lower activation threshold than mature DG cells. Furthermore, an enhanced plasticity and long-term potentiation (LTP) is detectable [47,48][47][48].
2. Major Depressive Disorder and Adult Neurogenesis
With increasing incidence and a high lifetime prevalence of 10–20% in the human population, MDD is one of the most studied psychiatric diseases [55][49]. MDD impacts mood and behavior, as well as various physical functions, such as appetite and sleep, and can lead to suicidal behavior. The causes for the development of the disease are multifactorial and not yet completely understood at the neurophysiological and molecular levels. Neuroendocrinological data hint towards a dysregulation of the HPA axis, since patients with hypercortisolism or exogenous glucocorticoid (GC) treatment more often develop MDD than healthy individuals [56][50]. Furthermore, the GC cortisol in humans and corticosterone in rodents are the most important stress hormones, highly elevated during periods of chronic stress and regarded as the main effector for the development of depression [31]. There is a variety of animal rodent models to mimic MDD symptoms, which basically consist of different stressors applied with distinct timing. In addition, also chronic corticosterone treatment induces a depressive-like phenotype in rodents. For an overview of animal stress models and depressive-like symptoms, see Table 1, and for further description of animal model protocols, a recent review [57][51].
Table 1. Summary of different chronic stress protocols in rodents, their behavioral outcome, and effect on adult hippocampal neurogenesis.
| Protocol of Stress | Behavior | Effect on Neurogenesis (↓ Decreased; ↔ Unchanged; ↑ Increased) |
|---|---|---|
| Chronic social stress, Chronic social defeat stress (CSDS) |
↑ Anhedonia, Social avoidance, ↑ Sleep disturbances, ↓ Exploratory anxiety, ↓ Weight | ↓ Simon et al., 2005; Schloesser et al., 2010; Jiang et al., 2019 [73,74,75][52][53][54] ↔ Hanson et al., 2011 [76][55] ↑ Lagace et al., 2010 [77][56] |
| Chronic unpredictable stress (CUS), (Unpredictable) Chronic mild stress ((U)CMS) |
↑ Anhedonia, ↑ Sleep disturbances, ↑ Behavioral despair, ↓ Grooming, ↓ Weight | ↓ Jayatissa et al., 2006, 2009; Toth et al., 2008; Surget et al., 2011; Dioli et al., 2017 [78,79,80,81,82][57][58][59][60][61] ↔ Lee et al., 2006 [83][62] |
| Chronic corticosteroid treatment | ↑ Anhedonia, ↑ Behavioral despair, ↑ Anxiety | ↓ Ekstrand et al., 2008; Brummelte and Galea, 2010; Pazini et al., 2017; Levone et al., 2020 [84,85,86,87][63][64][65][66] |
| Repeated restraint stress | ↑ Anhedonia, ↑ Anxiety, ↑ Behavioral despair | ↓ Luo et al., 2005; Rosenbrock et al., 2005; Snyder et al., 2011 [38,88,89][38][67][68] ↔ O’Leary et al., 2012 [90][69] ↑ Parihar et al., 2011 [91][70] |
| Early life stress (ELS) | ↑ Anhedonia, ↑ Anxiety, ↑ Behavioral despair, ↓ Learning, ↓ Locomotion | ↓ Mirescu et al., 2004; Kikusui et al., 2009; Lajud et al., 2012 [92,93,94][71][72][73] |
| Prenatal (restraint of pregnant dams) | ↑ Anhedonia, ↑ Anxiety, ↑ Behavioral despair | ↓ Lemaire et al., 2000; Bosch et al., 2006; Mandyam et al., 2008; Lucassen et al., 2009 [95,96,97,98][74][75][76][77] |
| Learned helplessness (chronic tail or footshocks) (LH) | ↓ Active avoidance, ↑ Sleep disturbances, ↓ Weight | ↓ Malberg and Duman, 2003 [99][78] ↔ Van der Borght et al., 2005 [100][79] |
| Social isolation (SI) | ↑ Anxiety, ↑ Behavioral despair, ↓ Learning, |
↓ Westenbroek et al., 2004; Spritzer et al., 2011; Chan et al., 2017 [101,[102,80103]][81][82] |
| Lipopolysaccharide-induced sickness behavior | ↑ Anhedonia, ↑ Lethargy, ↓ Appetite and food intake, ↑ Anxiety | ↓ Ekdahl et al., 2003; Monje, 2003; Yirmiya and Goshen, 2011; Perez-Dominguez et al., 2019 [104,105,106,107][83][84][85][86] ↔ Depino, 2015 [108][87] |
It is commonly known that the hippocampus is an important mediator of the negative feedback of the HPA axis involved in proper stress response [58][88]. Past studies, using postmortem analysis or magnetic resonance imaging (MRI), have revealed reductions in hippocampal volume of depressed patients [59,60][89][90]. Interestingly, in PTSD, a recent study reported a smaller human DG volume pretrauma as a predisposing vulnerability factor [61][91], which could also apply to MDD.
Like humans, rodents do not all develop depressive-like symptoms after chronic stress exposure, and hence, can be subdivided into resilient and susceptible groups based on their individual behavioral responses to stress [62][92]. Interestingly, this variation of the stress response can be linked to a reduction of hippocampal volume after CSDS in susceptible compared to non-stressed control mice [63][93]. Reductions of hippocampal volume could be either due to reduced neuroplasticity by dendritic growth arrest or atrophy leading to shortening of dendritic length and consequently to a reduction in spine density, which was observed in the CA3 region, and/or by the decreased generation of new neurons in the DG [64,65,66][94][95][96]. It is also unknown whether changes in adult neurogenesis and CA3 dendritic morphology are linked or are independent of each other, whereby one study in mice suggests that inhibiting adult neurogenesis for several months can lead to CA3 atrophy [67][97].
In rodent animal models, it is well established that protocols of chronic stress or chronic corticosterone treatment, used as a model of HPA axis overactivity, decrease adult neurogenesis (Table 1). Most, but not all, studies demonstrated deficits in neural stem/progenitor proliferation and/or differentiation, addressing also decreased cellular survival in the SGZ of the adult hippocampus (reviewed and discussed in Levone et al. (2015) [44]). Recent studies suggest that this might also be true for humans, by observing decreased numbers of granule cells in the DG of non-medicated depressed patients compared to healthy individuals and increased hippocampal neurogenesis and granule cell layer volume in antidepressant-treated compared to non-medicated patients [68,69,70][98][99][100]. In humans, early life adversity is one of the risk factors to develop MDD, including suicidal behavior in adulthood [71][101]. Interestingly, Boldrini et al. (2019) also demonstrated that an increased volume of DG is associated with resilience to early life adversity, presumably due to increased neurogenesis during childhood [72][102].
2.1. Antidepressants Acting on Adult Neurogenesis
It is most widely accepted that MDD patients display monoaminergic deficits [109[103][104],110], which are restored by treatment with the most common antidepressants targeting the serotonergic and norepinephrinergic systems. The majority of antidepressants need to be administered for at least six weeks to two months until full effectiveness, which opposes the impact of acute functioning. Rather a neuroplasticity-related mechanism is suggested, which seems to involve upregulation of brain-derived neurotrophic factor (BDNF) and thereby the antidepressant-induced enhancement of neurogenesis (see Section 3.1.3) [43,111][43][105]. As mentioned above, the full maturation of newly built hippocampal neurons takes approximately two months, and indeed, adult neural stem and precursor cells are positively regulated by serotonin (5-HT) [112][106] and norepinephrine [113,114,115][107][108][109]. In line with this, ablation studies with X-irradiation or cytostatic agents demonstrated that adult neurogenesis is necessary to ameliorate anxiety- and/or depressive-like behavioral effects exerted by antidepressants [116,117][110][111]. Moreover, a recent publication reports that selectively suppressing the excitability of newborn neurons by chemogenetic approaches without changing neurogenesis rate abolishes the antidepressant effect of the selective serotonin reuptake inhibitor (SSRI) fluoxetine, and that remarkably, activation of these neurons is sufficient to alleviate anxiety- and depressive-like behavior [46]. Not necessarily contrasting to this, other studies also demonstrated neurogenesis-independent mechanisms of antidepressants with a pivotal role in inducing remodeling of dendrites and synapses in mood-regulating limbic brain regions, which seems to account for an additional short-term effect of antidepressants [118,119,120][112][113][114].
Interestingly, a recent publication showed that blockade of indolamine 2, 3-dioxygenase 1 (IDO-1), an enzyme of the kynurenine pathway, associated with reduced 5-HT levels and hyperactivated in depression, ameliorated impaired hippocampal neurogenesis and depressive-like symptoms in mice, which underlines the importance of neurogenesis in the mechanistic action of monoamine-increasing antidepressants [111][105]. It is well known that approximately 30–40% of depressed patients are treatment-resistant by monotherapy with common antidepressants and do not achieve full remission of symptoms, even if medicated with an additional antidepressant after monotherapy [121][115]. Recent studies have shown that ketamine, an open channel blocker of the N-methyl-D-aspartate receptor (NMDAR), is effective for patients with treatment-resistant depression. Interestingly, similarly to monoaminergic antidepressants, also ketamine seems to act via augmented BDNF expression and a subsequent increase of adult neurogenesis, which was evident in the ventral hippocampus of adult mice [122][116]. In addition, electroconvulsive therapy (ECT), an efficient treatment for severe and refractory unipolar and bipolar depression, has remarkable antidepressant [123][117] and proneurogenic [124][118] properties. The subfield analysis of MRI scans showed that ECT in depressed patients increases the volume of major hippocampal regions and the DG [125,126][119][120]. Furthermore, the longitudinal analysis of hippocampal volume showed that hippocampal baseline is predictive of subsequent clinical outcomes [127][121]. Of note, the latter finding that is suggestive of increased neurogenesis is corroborated by studies with electroconvulsive stimulation (ECS), the analogous treatment for rodents, in animal models of depression. In mice treated with corticosterone (a stress model of depression, see Table 1), ECS significantly increased the number of newborn neurons, and more importantly, neurogenesis was required for the antidepressant effect of ECS, since mice lacking neurogenesis did not respond to the therapy [128][122]. Similar results were obtained in MAP6 knock-out (KO) mice, which share behavioral and neurobiological features of depression, including reduced neurogenesis and altered excitatory and monoaminergic transmission [129][123]. Interestingly, ECS in these mice not only improved neurogenesis and behavior, but also induced the expression of BDNF. Hence, different classes of antidepressants likely share the same cellular mechanism of action via restoration of adult neurogenesis by BDNF augmentation.
Whereas intact adult hippocampal neurogenesis certainly is required for antidepressant effects, a causative role for neurogenesis in depression is more difficult to be confirmed. Whether a reduction or ablation of adult neurogenesis alone is sufficient to induce depressive-like symptoms is still a controversy, due to contradicting results of diverse studies, which have been extensively discussed elsewhere [40,110,130[40][104][124][125],131], and will be taken up in Section 5. Nevertheless, since the increase of adult neurogenesis is sufficient to reduce anxiety- and depression-like behaviors [41[41][42],42], a positive role of adult neurogenesis in stress-related resilient behavior seems very likely.
2.2. MDD and Dysregulated Immune System
Together with HPA axis overactivation and monoamine dysfunction, dysregulated immune response has been implicated in the pathogenesis of MDD [132,133][126][127]. An unbalance between the adaptive and the innate immune response has emerged as a typical immunological signature of MDD [134][128]. While the number of activated monocytes is increased, T lymphocytes are reduced [135,136][129][130]. Consistent with the monocyte activation, circulating levels of proinflammatory cytokines, such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6), have been shown to be increased in patients with MDD [137][131] and with PTSD [138][132], and in animal models of stress [139,140][133][134]. For this reason, cytokine serum levels have been proposed as reliable biomarkers for both MDD and PTSD [138,141][132][135]. Noteworthy, both preclinical and clinical studies point to IL-6 as a reliable predictive marker of MDD susceptibility levels. Indeed, higher levels of IL-6 in childhood, likely because of adverse events [142][136], have been associated with increased risk of depression in adulthood [143][137] and shown to predict stress resilience in animal models of chronic stress [139][133]. The pathogenic role of immune dysfunction in MDD is further supported by the results of a large meta-analysis showing that a history of infections or autoimmune diseases is a risk factor for MDD [144][138].
Hence, it appears that a proinflammatory milieu might be a predisposing factor for later development of MDD and MDD-related suppression of neurogenesis. Proinflammatory cytokines might affect neurogenesis by binding their receptors expressed on both NPCs and neurons, thereby directly regulating NPC fate or by modulating the synaptic inputs onto NPCs, respectively [145][139]. Indirect mechanisms might also be existing and might arise by the complex relationship between the immune system and HPA axis and the 5-HT biosynthetic pathways, which, as already mentioned, directly modulate neurogenesis. Indeed, consistent with the MDD neuroendocrine and immunological picture, proinflammatory cytokines stimulate GCs and are regulated by GCs [146][140]. Moreover, proinflammatory cytokines can reduce tryptophan availability in the gut, thus impairing gut microbiota-mediated biosynthesis of 5-HT precursor [147][141].
3. Modulation of Neurogenesis
3.1. Positive Modulation of Adult Neurogenesis by Potential Resilience Factors
A variety of factors or conditions upregulating adult hippocampal neurogenesis rate have also been described independently of neurogenesis to be “resilience factors” or to act in an antidepressant manner. This means that mechanistically they could modulate adult neurogenesis to promote stress resilience. In the following, we will summarize what is known about some prominent regulatory factors, such as BDNF and endocannabinoids (eCBs), or conditions, such as exercise and enriched environment in the context of stress resilience by regulating adult hippocampal neurogenesis. In addition, negative modulation of neurogenesis by stress and its disease-promoting role will be delineated (Figure 1).
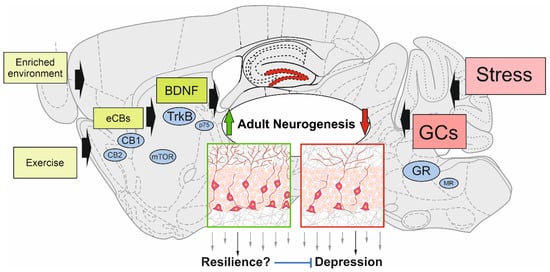
Figure 1. Schematic view of modulating factors of adult hippocampal neurogenesis. Enriched environment, exercise, and molecular players (e.g., endocannabinoids (eCBs) and brain-derived neurotrophic factor (BDNF)) have the potential to upregulate the generation of adult-born neurons in the dentate gyrus. This could confer resilience to the development of depressive-like symptoms through the stress-related decline of adult neurogenesis induced by glucocorticoids (GCs). The main signaling pathways of positive modulators and stress are depicted: cannabinoid receptor type-1 and -2 (CB1; CB2); mammalian target of rapamycin (mTOR); tropomyosin receptor kinase B (TrkB); p75 neurotrophin receptor (p75); glucocorticoid receptor (GR); mineralocorticoid receptor (MR).
3.1.1. Environmental Enrichment (EE) and Physical Exercise (PE)
EE and PE are convincingly associated with a broad spectrum of beneficial effects on the hippocampus, including boosting neurogenesis [148][142]. In animal studies, EE refers to an experimental setting in which rodents are kept in a larger group and in the presence of multiple objects (toys, nesting material, running wheel), to provide animals with social, physical, and cognitive stimulation [149][143]. PE usually refers to running, mainly performed on a running wheel, to mimic an aerobic activity. Both experimental paradigms are intended to simulate enhanced cognitive and physical stimuli in humans. Despite the lack of direct evidence of improved neurogenesis in humans, an increase of hippocampal volume and cerebral blood flow in this region in people engaged in exercise are reasonably considered suggestive of potentiated neurogenesis in the DG [150][144]. Pioneering studies published in the late ninetieth of the last century showed that running and EE increase the number of proliferating neurons in the DG [23[23][145][146],151,152], paving the way for flourishing literature on this topic, as nicely reviewed elsewhere [148,149,150,153][142][143][144][147]. Both paradigms have been shown to influence several aspects of neurogenesis, such as proliferation [154][148], maturation and morphology [155][149], and functional integration of newborn neurons [156][150], which contribute to increased synaptic plasticity in the DG area [151][145] and improved spatial memory [157][151]. Acute bout (few days) of running were shown to induce a fast increase of the number of proliferating neurons with prosurvival effects of the progeny [154][148]. Interestingly, exercise was proven to be the neurogenic component of EE [158,159,160][152][153][154] and to improve neurogenesis even in old animals, counteracting the age- and pathological-dependent neurogenesis reduction [150,154,161][144][148][155].
Beyond the peripheral muscle- and endocrine-derived factors, central nervous system (CNS) intrinsic mechanisms have been claimed to play a role in the exercise-mediated proneurogenic effects. Among these, experience-driven increased glutamatergic activity, and upregulation of BDNF levels and signaling are the most accountable [162][156].
3.1.2. Endocannabinoids (eCBs)
Endocannabinoids (eCBs) are signaling molecules synthesized from membrane lipid components and are derivatives of arachidonic acid, forming the two major eCBs 2-arachidonoyl glycerol (2-AG), and arachidonoyl ethanol amide (also called anandamide, AEA). The high lipophilicity prevents storage in vesicles, and therefore, the intensity of eCB signaling is driven by the activities of the eCB synthesizing and degrading enzymes. eCBs can act in an autocrine and paracrine manner, and are ligands for different receptors, whereby the major receptors are the cannabinoid type 1 receptor (CB1) and type 2 receptor (CB2) [163][157]. Yet, AEA can also activate TRPV1 (transient receptor potential cation channel subfamily V member 1), while 2-AG also stimulates the GABAA receptor [164,165][158][159]. The eCB signaling is involved in many physiological and pathophysiological processes both in the CNS and in peripheral organs [164][158]. In the context of adult neurogenesis, the research has focused on CB1 and CB2. While these receptors and eCB synthesizing and degrading enzymatic machinery have been reported to be present in NPCs in the SVZ of the adult hippocampus [25[25][160],166], the intensity and exact mode of eCB signaling in NPC or onto NPC are difficult to be determined. As these eCB components are additionally expressed in cells surrounding the neurogenic niches in the SGZ, the functionality of eCB signaling regarding the regulation of adult neurogenesis is complex and may act in a paracrine and/or autocrine manner onto neural stem cells and NPCs.
It has been reported that, in general, eCB signaling, as well as phytocannabinoids regulate adult neurogenesis positively, mostly via CB1 and CB2 [166[160][161],167], possibly through multiple mechanisms, including proliferation, antiapoptotic defense, antioxidant defense, immunoregulation, and autophagy/mitophagy [166][160]. Most of the investigations have addressed these functions under physiological conditions, and only a few investigations addressed stimulated conditions, with positive (e.g., exercise) or negative (e.g., stress) annotation. As discussed above, a link between antidepressant intervention and adult neurogenesis has frequently been reported. In fact, in a mouse model of depressive-like behavior by using CUS, the inhibition of the 2-AG degrading enzyme monoacyl glycerol lipase (MAGL) by chronic application of JZL184 prevented the CUS-induced increase of feeding latency in the novelty-induced suppression of feeding, and immobility time in the forced swim test [168][162]. The positive behavioral outcome went along with the prevention of CUS-induced impaired adult neurogenesis in the SGZ, and a form of LTP in the DG known to be neurogenesis-dependent. These effects were associated with the normalization of CUS-induced decrease of mTOR (mammalian target of rapamycin) [169][163]. The mTOR signaling pathway was shown to be compromised in MDD subjects [170][164], whereas mTOR activation acts in an antidepressant manner [171][165]. Along with this, activation of mTOR signaling is known to play pivotal roles in adult neural stem cell regulation by particularly upregulating proliferation of the transient amplifying stem cell pool [172][166], but also by impacting NPC differentiation (for review, see the work by the authors of [173][167]). A recent other investigation addressed the influence of the microbiome on the eCB system and adult neurogenesis [174][168]. In an elegant set of experiments using unpredictable chronic mild stress (UCMS) as a mouse model of depression, and fecal microbiota transfer from these mice to non-UCMS mice, the authors rescued the microbiota-transmitted depressive-like behavior by pharmacological inhibition of MAGL with JZL184, concomitantly with the restoration of adult neurogenesis. Furthermore, it was also shown that complementation of UCMS microbiota with Lactobacillaceae alleviated depressive-like symptoms and restored neurogenesis levels in recipients of UCMS microbiota.
As outlined above, exercise is an efficient intervention for increasing adult neurogenesis. Pharmacological blockade of the CB1 alleviated the exercise-induced increase in proliferation in the SGZ [175][169]. In another study, though, using CB1 deficient mice, such a CB1 dependency on neurogenesis was not observed upon a 6-week running period, but the CB1 deficient mice showed reduced motivation to run [176][170]. The reasons for these divergent observations have not been clarified.
In summary, the current data on the involvement of the eCB system in stress coping and neurogenesis suggest that the enhancement of eCB signaling, in particular 2-AG, is beneficial for alleviating stress-induced depressive-like behavior, and concomitantly, to the stress-induced blunting of adult neurogenesis. The underlying mechanisms of the stimulatory effects on neurogenesis have still to be further investigated.
3.1.3. Brain-Derived Neurotrophic Factor (BDNF)
The neurotrophin BDNF regulates survival, proliferation, differentiation, and migration of neural stem and progenitor cells in vitro and in vivo during neural development of the embryo, as well as in adult neurogenesis [177,178,179,180][171][172][173][174]. In mature neurons, BDNF is also well known for its function in synaptic plasticity and LTP formation, thereby controlling cognition, learning, and memory, but also mood [43,181,182,183][43][175][176][177]. BDNF is secreted at the pre- and postsynaptic side either as proprotein or mature BDNF in an activity-dependent manner or by the constitutive pathway of exocytosis [184,185,186][178][179][180]. BDNF exerts its functions through binding to its two receptors, the high affinity tropomyosin receptor kinase B (TrkB) and the low-affinity p75 pan neurotrophin receptor (p75NTR). Besides being expressed on the vast majority of neurons, the occurrence of both receptor types has been demonstrated in both adult neurogenic niches exhibiting dynamic expression during distinct stages of adult neurogenesis [187,188][181][182]. BDNF signaling through the TrkB receptor acts mainly via the PI3K/Akt pathway to positively regulate cellular survival and structural plasticity, whereas the MAP kinase pathway in concert with PLCγ is the main player in regulating cellular proliferation and differentiation. Binding to p75NTR was demonstrated to have opposing functions, e.g., the reduction of dendritic arborization, apoptosis, and long-term depression, also reflecting the enhanced binding of pro-BDNF, for which opposing physiological roles have been demonstrated [189,190,191,192][183][184][185][186].
Role of BDNF in MDD
It has been widely shown that serum BDNF availability correlates with mood changes and reflects the pathophysiological state in mood disorders, as well as with structural changes in specific brain regions, such as the hippocampus and cortical areas [193,194,195,196,197][187][188][189][190][191]. Moreover, BDNF serum levels seem to reflect BDNF brain levels [198][192]. Altogether this implicates BDNF as a potential biomarker for MDD, but also for other mood disorders [199][193]. Indeed, recently, also DNA-methylation profiles of the BDNF promoter were suggested as MDD biomarker, because depressed and healthy individuals could be clearly classified into two groups by this epigenetic modification [200][194]. The BDNF hypothesis of depression is justified because opposing actions of stress and antidepressant treatment are observed on existing BDNF levels in serum and limbic brain regions, such as the hippocampus [182][176]. Stress significantly suppresses mRNA and protein BDNF levels in the hippocampus, particularly in the DG and CA3 hippocampal subfields, and thereby impairs downstream targets of signaling pathways implicated in neuroplasticity [201,202][195][196]. Two important meta-analyses could directly prove decreased serum BDNF levels in depressed, suicidal patients, whereas BDNF was increased after antidepressant treatment in humans [195,196][189][190]. The question of how BDNF exerts its antidepressant effect is still not fully understood, since the regulation by BDNF could appear at the level of neuronal excitability, as well as regarding the regulation of adult neurogenesis or both. Furthermore, brain atrophy caused by stress [203][197] could be potentially counteracted by BDNF, serving as a survival factor for degenerating neurons. However, this last point is unlikely because some antidepressants reported an increase of BDNF that did not reverse stress-induced atrophy [182,203][176][197].
3.2. Negative Modulation of Adult Neurogenesis by Stress
Long-term exposure to environmental, physical, and psychosocial stress is a recognized risk factor for MDD, also referred to as stress-related disorder [132][126]. A plethora of stressors contributes to the development of MDD, including traumatic events, such as bereavement, repetitive job hassles, diagnosis of a disabling disease, physical or sexual abuse. The time-window of trauma exposure has a leading role in determining the body’s structural and functional changes in response to stress. In this respect, early life stress (ELS), such as childhood trauma (for example, abuse), lack of maternal care, poor nutritional intake, triggers significant changes in the brain with psychological consequences in adulthood [226][198]. The hippocampus, which mostly develops postnatally in both humans and rodents [227[199][200],228], is highly sensitive to precocious stress. ELS in rodents was shown to impair adult neurogenesis, in correlation with impaired learning and memory functions (reviewed by the authors of [226][198]) specifically in male rodents [229[201][202],230], reviewed by the authors of [231][203].
From a neuroendocrine point of view, acute stress engages a fast and self-limiting body reaction that implicates the involvement of the stress hormones, cortisol, norepinephrine, and epinephrine, the immune system, and stress-sensitive brain areas, such as the hippocampus. The complex interaction among these factors underlying the so-called “fight or flight response” is a beneficial protective mechanism that prepares the body to react to stressors [232][204]. A crucial role in the stress system is played by GCs and the HPA axis. Activation of the HPA axis starting from the release of corticotropin-releasing hormone (CRH) from the hypothalamus to stimulate the pituitary release of adrenocorticotropin hormone (ACTH) leads to the final synthesis and release of cortisol in humans and corticosterone in rodents from adrenal glands [146][140]. GC levels, in turn, block the HPA axis, through negative feedback over the hypothalamus, and as mentioned above, the hippocampus. This area is particularly rich in GC receptor (GR), which, in contrast to the other GC responsive receptor, the mineralocorticoid receptor (MR), has been implicated in the negative feedback to stress [233][205].
References
- Kuhn, H.G.; Toda, T.; Gage, F.H. Adult hippocampal neurogenesis: A coming-of-age story. J. Neurosci. 2018, 38, 10401–10410.
- Snyder, J.S. Questioning human neurogenesis. Nature 2018, 555, 315–316.
- Gandhi, S.; Gupta, J.; Tripathi, P.P. The Curious Case of Human Hippocampal Neurogenesis. ACS Chem. Neurosci. 2019, 10, 1131–1132.
- Moreno-Jiménez, E.P.; Terreros-Roncal, J.; Flor-García, M.; Rábano, A.; Llorens-Martín, M. Evidences for Adult Hippocampal Neurogenesis in Humans. J. Neurosci. 2021, 41, 2541–2553.
- Tobin, M.K.; Musaraca, K.; Disouky, A.; Shetti, A.; Bheri, A.; Honer, W.G.; Kim, N.; Dawe, R.J.; Bennett, D.A.; Arfanakis, K.; et al. Human Hippocampal Neurogenesis Persists in Aged Adults and Alzheimer’s Disease Patients. Cell Stem Cell 2019, 24, 974–982.e3.
- Boldrini, M.; Fulmore, C.A.; Tartt, A.N.; Simeon, L.R.; Pavlova, I.; Poposka, V.; Rosoklija, G.B.; Stankov, A.; Arango, V.; Dwork, A.J.; et al. Human Hippocampal Neurogenesis Persists throughout Aging. Cell Stem Cell 2018, 22, 589–599.e5.
- Spalding, K.L.; Bergmann, O.; Alkass, K.; Bernard, S.; Salehpour, M.; Huttner, H.B.; Boström, E.; Westerlund, I.; Vial, C.; Buchholz, B.A.; et al. XDynamics of hippocampal neurogenesis in adult humans. Cell 2013, 153, 1219.
- Eriksson, P.S.; Perfilieva, E.; Björk-Eriksson, T.; Alborn, A.M.; Nordborg, C.; Peterson, D.A.; Gage, F.H. Neurogenesis in the adult human hippocampus. Nat. Med. 1998, 4, 1313–1317.
- Knoth, R.; Singec, I.; Ditter, M.; Pantazis, G.; Capetian, P.; Meyer, R.P.; Horvat, V.; Volk, B.; Kempermann, G. Murine features of neurogenesis in the human hippocampus across the lifespan from 0 to 100 years. PLoS ONE 2010, 5, e8809.
- Moreno-Jiménez, E.P.; Flor-García, M.; Terreros-Roncal, J.; Rábano, A.; Cafini, F.; Pallas-Bazarra, N.; Ávila, J.; Llorens-Martín, M. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 2019, 25, 554–560.
- Sorrells, S.F.; Paredes, M.F.; Zhang, Z.; Kang, G.; Pastor-Alonso, O.; Biagiotti, S.; Page, C.E.; Sandoval, K.; Knox, A.; Connolly, A.; et al. Positive Controls in Adults and Children Support That Very Few, If Any, New Neurons Are Born in the Adult Human Hippocampus. J. Neurosci. 2021, 41, 2554–2565.
- Sorrells, S.F.; Paredes, M.F.; Cebrian-Silla, A.; Sandoval, K.; Qi, D.; Kelley, K.W.; James, D.; Mayer, S.; Chang, J.; Auguste, K.I.; et al. Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature 2018, 555, 377–381.
- Cipriani, S.; Ferrer, I.; Aronica, E.; Kovacs, G.G.; Verney, C.; Nardelli, J.; Khung, S.; Delezoide, A.L.; Milenkovic, I.; Rasika, S.; et al. Hippocampal radial glial subtypes and their neurogenic potential in human fetuses and healthy and Alzheimer’s disease adults. Cereb. Cortex 2018, 28, 2458–2478.
- Lucassen, P.J.; Fitzsimons, C.P.; Salta, E.; Maletic-Savatic, M. Adult neurogenesis, human after all (again): Classic, optimized, and future approaches. Behav. Brain Res. 2020, 381, 112458.
- Lucassen, P.J.; Toni, N.; Kempermann, G.; Frisen, J.; Gage, F.H.; Swaab, D.F. Limits to human neurogenesis—Really? Mol. Psychiatry 2020, 25, 2207–2209.
- Kempermann, G.; Gage, F.H.; Aigner, L.; Song, H.; Curtis, M.A.; Thuret, S.; Kuhn, H.G.; Jessberger, S.; Frankland, P.W.; Cameron, H.A.; et al. Human Adult Neurogenesis: Evidence and Remaining Questions. Cell Stem Cell 2018, 23, 25–30.
- Denoth-Lippuner, A.; Jessberger, S. Formation and integration of new neurons in the adult hippocampus. Nat. Rev. Neurosci. 2021, 22, 223–236.
- Obernier, K.; Alvarez-Buylla, A. Neural stem cells: Origin, heterogeneity and regulation in the adult mammalian brain. Development 2019, 146, 156059.
- Zhao, C.; Deng, W.; Gage, F.H. Mechanisms and Functional Implications of Adult Neurogenesis. Cell 2008, 132, 645–660.
- Imayoshi, I.; Sakamoto, M.; Ohtsuka, T.; Takao, K.; Miyakawa, T.; Yamaguchi, M.; Mori, K.; Ikeda, T.; Itohara, S.; Kageyama, R. Roles of continuous neurogenesis in the structural and functional integrity of the adult forebrain. Nat. Neurosci. 2008, 11, 1153–1161.
- Ben Abdallah, N.M.B.; Filipkowski, R.K.; Pruschy, M.; Jaholkowski, P.; Winkler, J.; Kaczmarek, L.; Lipp, H.P. Impaired long-term memory retention: Common denominator for acutely or genetically reduced hippocampal neurogenesis in adult mice. Behav. Brain Res. 2013, 252, 275–286.
- Ben Abdallah, N.M.B.; Slomianka, L.; Vyssotski, A.L.; Lipp, H.P. Early age-related changes in adult hippocampal neurogenesis in C57 mice. Neurobiol. Aging 2010, 31, 151–161.
- Kempermann, G.; Kuhn, H.G.; Gage, F.H. More hippocampal neurons in adult mice living in an enriched environment. Nature 1997, 386, 493–495.
- Drapeau, E.; Mayo, W.; Aurousseau, C.; Le Moal, M.; Piazza, P.V.; Abrous, D.N. Spatial memory performances of aged rats in the water maze predict levels of hippocampal neurogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 14385–14390.
- Zimmermann, T.; Maroso, M.; Beer, A.; Baddenhausen, S.; Ludewig, S.; Fan, W.; Vennin, C.; Loch, S.; Berninger, B.; Hofmann, C.; et al. Neural stem cell lineage-specific cannabinoid type-1 receptor regulates neurogenesis and plasticity in the adult mouse hippocampus. Cereb. Cortex 2018, 28, 4454–4471.
- Fanselow, M.S.; Dong, H.W. Are the Dorsal and Ventral Hippocampus Functionally Distinct Structures? Neuron 2010, 65, 7–19.
- Kheirbek, M.A.; Drew, L.J.; Burghardt, N.S.; Costantini, D.O.; Tannenholz, L.; Ahmari, S.E.; Zeng, H.; Fenton, A.A.; Hen, R. Differential Control of Learning and Anxiety along the Dorsoventral Axis of the Dentate Gyrus. Neuron 2013, 77, 955–968.
- Kheirbek, M.A.; Hen, R. Dorsal vs. Ventral Hippocampal Neurogenesis: Implications for Cognition and Mood. Neuropsychopharmacology 2011, 36, 373–374.
- Komorowski, R.W.; Garcia, C.G.; Wilson, A.; Hattori, S.; Howard, M.W.; Eichenbaum, H. Ventral Hippocampal Neurons Are Shaped by Experience to Represent Behaviorally Relevant Contexts. J. Neurosci. 2013, 33, 8079–8087.
- Huckleberry, K.A.; Shue, F.; Copeland, T.; Chitwood, R.A.; Yin, W.; Drew, M.R. Dorsal and ventral hippocampal adult-born neurons contribute to context fear memory. Neuropsychopharmacology 2018, 43, 2487–2496.
- Joëls, M. Corticosteroids and the brain. J. Endocrinol. 2018, 238, R121–R130.
- Cathomas, F.; Murrough, J.W.; Nestler, E.J.; Han, M.H.; Russo, S.J. Neurobiology of Resilience: Interface between Mind and Body. Biol. Psychiatry 2019, 86, 410–420.
- Kheirbek, M.A.; Klemenhagen, K.C.; Sahay, A.; Hen, R. Neurogenesis and generalization: A new approach to stratify and treat anxiety disorders. Nat. Neurosci. 2012, 15, 1613–1620.
- Frankland, P.W.; Köhler, S.; Josselyn, S.A. Hippocampal neurogenesis and forgetting. Trends Neurosci. 2013, 36, 497–503.
- Miller, S.M.; Sahay, A. Functions of adult-born neurons in hippocampal memory interference and indexing. Nat. Neurosci. 2019.
- Anacker, C.; Hen, R. Adult hippocampal neurogenesis and cognitive flexibility—linking memory and mood. Nat. Rev. Neurosci. 2017, 18, 335–346.
- Glover, L.R.; Schoenfeld, T.J.; Karlsson, R.-M.; Bannerman, D.M.; Cameron, H.A. Ongoing neurogenesis in the adult dentate gyrus mediates behavioral responses to ambiguous threat cues. PLoS Biol. 2017, 15, e2001154.
- Snyder, J.S.; Soumier, A.; Brewer, M.; Pickel, J.; Cameron, H.A. Adult hippocampal neurogenesis buffers stress responses and depressive behaviour. Nature 2011, 476, 458–462.
- Revest, J.M.; Dupret, D.; Koehl, M.; Funk-Reiter, C.; Grosjean, N.; Piazza, P.V.; Abrous, D.N. Adult hippocampal neurogenesis is involved in anxiety-related behaviors. Mol. Psychiatry 2009, 14, 959–967.
- Yun, S.; Donovan, M.H.; Ross, M.N.; Richardson, D.R.; Reister, R.; Farnbauch, L.A.; Fischer, S.J.; Riethmacher, D.; Gershenfeld, H.K.; Lagace, D.C.; et al. Stress-induced anxiety- and depressive-like phenotype associated with transient reduction in neurogenesis in adult Nestin-CreERT2/diphtheria toxin fragment A transgenic mice. PLoS ONE 2016, 11, e0147256.
- Hill, A.S.; Sahay, A.; Hen, R. Increasing Adult Hippocampal Neurogenesis is Sufficient to Reduce Anxiety and Depression-Like Behaviors. Neuropsychopharmacology 2015, 40, 2368–2378.
- Eliwa, H.; Brizard, B.; Le Guisquet, A.M.; Hen, R.; Belzung, C.; Surget, A. Adult neurogenesis augmentation attenuates anhedonia and HPA axis dysregulation in a mouse model of chronic stress and depression. Psychoneuroendocrinology 2021, 124.
- Park, S.C. Neurogenesis and antidepressant action. Cell Tissue Res. 2019, 377, 95–106.
- Levone, B.R.; Cryan, J.F.; O’Leary, O.F. Role of adult hippocampal neurogenesis in stress resilience. Neurobiol. Stress 2015, 1, 147–155.
- Anacker, C.; Luna, V.M.; Stevens, G.S.; Millette, A.; Shores, R.; Jimenez, J.C.; Chen, B.; Hen, R. Hippocampal neurogenesis confers stress resilience by inhibiting the ventral dentate gyrus. Nature 2018, 559, 98–102.
- Tunc-Ozcan, E.; Peng, C.Y.; Zhu, Y.; Dunlop, S.R.; Contractor, A.; Kessler, J.A. Activating newborn neurons suppresses depression and anxiety-like behaviors. Nat. Commun. 2019, 10, 1–9.
- Ge, S.; Yang, C.H.; Hsu, K.S.; Ming, G.L.; Song, H. A Critical Period for Enhanced Synaptic Plasticity in Newly Generated Neurons of the Adult Brain. Neuron 2007.
- Tannenholz, L.; Hen, R.; Kheirbek, M.A. GluN2B-Containg NMDA Receptors on Adult-Born Granule Cells Contribute to the Antidepressant Action of Fluoxetine. Front. Neurosci. 2016, 10.
- Ferrari, A.J.; Somerville, A.J.; Baxter, A.J.; Norman, R.; Patten, S.B.; Vos, T.; Whiteford, H.A. Global variation in the prevalence and incidence of major depressive disorder: A systematic review of the epidemiological literature. Psychol. Med. 2013, 43, 471–481.
- Starkman, M.N.; Giordani, B.; Gebarski, S.S.; Berent, S.; Schork, M.A.; Schteingart, D.E. Decrease in cortisol reverses human hippocampal atrophy following treatment of Cushing’s disease. Biol. Psychiatry 1999, 46, 1595–1602.
- Dudek, K.A.; Dion-Albert, L.; Kaufmann, F.N.; Tuck, E.; Lebel, M.; Menard, C. Neurobiology of resilience in depression: Immune and vascular insights from human and animal studies. Eur. J. Neurosci. 2021, 53, 183–221.
- Simon, M.; Czéh, B.; Fuchs, E. Age-dependent susceptibility of adult hippocampal cell proliferation to chronic psychosocial stress. Brain Res. 2005, 1049, 244–248.
- Schloesser, R.J.; Lehmann, M.; Martinowich, K.; Manji, H.K.; Herkenham, M. Environmental enrichment requires adult neurogenesis to facilitate the recovery from psychosocial stress. Mol. Psychiatry 2010, 15, 1152–1163.
- Jiang, N.; Lv, J.-W.; Wang, H.X.; Lu, C.; Wang, Q.; Xia, T.-J.; Bao, Y.; Li, S.-S.; Liu, X.M. Dammarane sapogenins alleviates depression-like behaviours induced by chronic social defeat stress in mice through the promotion of the BDNF signalling pathway and neurogenesis in the hippocampus. Brain Res. Bull. 2019, 153, 239–249.
- Hanson, N.D.; Owens, M.J.; Boss-Williams, K.A.; Weiss, J.M.; Nemeroff, C.B. Several stressors fail to reduce adult hippocampal neurogenesis. Psychoneuroendocrinology 2011, 36, 1520–1529.
- Lagace, D.C.; Donovan, M.H.; Decarolis, N.A.; Farnbauch, L.A.; Malhotra, S.; Berton, O.; Nestler, E.J.; Krishnan, V.; Eisch, A.J. Adult hippocampal neurogenesis is functionally important for stress-induced social avoidance. Proc. Natl. Acad. Sci. USA 2010, 107, 4436–4441.
- Jayatissa, M.N.; Bisgaard, C.; Tingström, A.; Papp, M.; Wiborg, O. Hippocampal cytogenesis correlates to escitalopram-mediated recovery in a chronic mild stress rat model of depression. Neuropsychopharmacology 2006, 31, 2395–2404.
- Jayatissa, M.N.; Henningsen, K.; West, M.J.; Wiborg, O. Decreased cell proliferation in the dentate gyrus does not associate with development of anhedonic-like symptoms in rats. Brain Res. 2009, 1290, 133–141.
- Toth, E.; Gersner, R.; Wilf-Yarkoni, A.; Raizel, H.; Dar, D.E.; Richter-Levin, G.; Levit, O.; Zangen, A. Age-dependent effects of chronic stress on brain plasticity and depressive behavior. J. Neurochem. 2008, 107, 522–532.
- Surget, A.; Tanti, A.; Leonardo, E.D.; Laugeray, A.; Rainer, Q.; Touma, C.; Palme, R.; Griebel, G.; Ibarguen-Vargas, Y.; Hen, R.; et al. Antidepressants recruit new neurons to improve stress response regulation. Mol. Psychiatry 2011, 16, 1177–1188.
- Dioli, C.; Patrício, P.; Trindade, R.; Pinto, L.G.; Silva, J.M.; Morais, M.; Ferreiro, E.; Borges, S.; Mateus-Pinheiro, A.; Rodrigues, A.J.; et al. Tau-dependent suppression of adult neurogenesis in the stressed hippocampus. Mol. Psychiatry 2017, 22, 1110–1118.
- Lee, K.J.; Kim, S.J.; Kim, S.W.; Choi, S.H.; Shin, Y.C.; Park, S.H.; Moon, B.H.; Cho, E.; Lee, M.S.; Choi, S.H.; et al. Chronic mild stress decreases survival, but not proliferation, of new-born cells in adult rat hippocampus. Exp. Mol. Med. 2006, 38, 44–54.
- Brummelte, S.; Galea, L.A.M. Chronic high corticosterone reduces neurogenesis in the dentate gyrus of adult male and female rats. Neuroscience 2010, 168, 680–690.
- Ekstrand, J.; Hellsten, J.; Wennström, M.; Tingström, A. Differential inhibition of neurogenesis and angiogenesis by corticosterone in rats stimulated with electroconvulsive seizures. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2008, 32, 1466–1472.
- Levone, B.R.; Codagnone, M.G.; Moloney, G.M.; Nolan, Y.M.; Cryan, J.F.; O’Leary, O.F. Adult-born neurons from the dorsal, intermediate, and ventral regions of the longitudinal axis of the hippocampus exhibit differential sensitivity to glucocorticoids. Mol. Psychiatry 2020.
- Pazini, F.L.; Cunha, M.P.; Azevedo, D.; Rosa, J.M.; Colla, A.; de Oliveira, J.; Ramos-Hryb, A.B.; Brocardo, P.S.; Gil-Mohapel, J.; Rodrigues, A.L.S. Creatine Prevents Corticosterone-Induced Reduction in Hippocampal Proliferation and Differentiation: Possible Implication for Its Antidepressant Effect. Mol. Neurobiol. 2017, 54, 6245–6260.
- Luo, C.; Xu, H.; Li, X.M. Quetiapine reverses the suppression of hippocampal neurogenesis caused by repeated restraint stress. Brain Res. 2005, 1063, 32–39.
- Rosenbrock, H.; Koros, E.; Bloching, A.; Podhorna, J.; Borsini, F. Effect of chronic intermittent restraint stress on hippocampal expression of marker proteins for synaptic plasticity and progenitor cell proliferation in rats. Brain Res. 2005, 1040, 55–63.
- O’Leary, O.F.; O’Connor, R.M.; Cryan, J.F. Lithium-induced effects on adult hippocampal neurogenesis are topographically segregated along the dorso-ventral axis of stressed mice. Neuropharmacology 2012, 62, 247–255.
- Parihar, V.K.; Hattiangady, B.; Kuruba, R.; Shuai, B.; Shetty, A.K. Predictable chronic mild stress improves mood, hippocampal neurogenesis and memory. Mol. Psychiatry 2011, 16, 171–183.
- Kikusui, T.; Ichikawa, S.; Mori, Y. Maternal deprivation by early weaning increases corticosterone and decreases hippocampal BDNF and neurogenesis in mice. Psychoneuroendocrinology 2009, 34, 762–772.
- Lajud, N.; Roque, A.; Cajero, M.; Gutiérrez-Ospina, G.; Torner, L. Periodic maternal separation decreases hippocampal neurogenesis without affecting basal corticosterone during the stress hyporesponsive period, but alters HPA axis and coping behavior in adulthood. Psychoneuroendocrinology 2012, 37, 410–420.
- Mirescu, C.; Peters, J.D.; Gould, E. Early life experience alters response of adult neurogenesis to stress. Nat. Neurosci. 2004, 7, 841–846.
- Lucassen, P.J.; Bosch, O.J.; Jousma, E.; Krömer, S.A.; Andrew, R.; Seckl, J.R.; Neumann, I.D. Prenatal stress reduces postnatal neurogenesis in rats selectively bred for high, but not low, anxiety: Possible key role of placental 11β-hydroxysteroid dehydrogenase type 2. Eur. J. Neurosci. 2009, 29, 97–103.
- Lemaire, V.; Koehl, M.; Le Moal, M.; Abrous, D.N. Prenatal stress produces learning deficits associated with an inhibition of neurogenesis in the hippocampus. Proc. Natl. Acad. Sci. USA 2000, 97, 11032–11037.
- Mandyam, C.D.; Crawford, E.F.; Eisch, A.J.; Rivier, C.L.; Richardson, H.N. Stress experiencedin utero reduces sexual dichotomies in neurogenesis, microenvironment, and cell death in the adult rat hippocampus. Dev. Neurobiol. 2008, 68.
- Bosch, O.J.; Krömer, S.A.; Neumann, I.D. Prenatal stress: Opposite effects on anxiety and hypothalamic expression of vasopressin and corticotropin-releasing hormone in rats selectively bred for high and low anxiety. Eur. J. Neurosci. 2006, 23, 541–551.
- Malberg, J.E.; Duman, R.S. Cell proliferation in adult hippocampus is decreased by inescapable stress: Reversal by fluoxetine treatment. Neuropsychopharmacology 2003, 28, 1562–1571.
- Van Der Borght, K.; Meerlo, P.; Paul, P.G.; Eggen, B.J.L.; Zee, E.A.V. Der Effects of active shock avoidance learning on hippocampal neurogenesis and plasma levels of corticosterone. Behav. Brain Res. 2005, 157, 23–30.
- Chan, J.N.M.; Lee, J.C.D.; Lee, S.S.P.; Hui, K.K.Y.; Chan, A.H.L.; Fung, T.K.H.; Sánchez-Vidaña, D.I.; Lau, B.W.M.; Ngai, S.P.C. Interaction effect of social isolation and high dose corticosteroid on neurogenesis and emotional behavior. Front. Behav. Neurosci. 2017, 11.
- Spritzer, M.D.; Ibler, E.; Inglis, W.; Curtis, M.G. Testosterone and social isolation influence adult neurogenesis in the dentate gyrus of male rats. Neuroscience 2011, 195, 180–190.
- Westenbroek, C.; Den Boer, J.A.; Veenhuis, M.; Ter Horst, G.J. Chronic stress and social housing differentially affect neurogenesis in male and female rats. Brain Res. Bull. 2004, 64, 303–308.
- Ekdahl, C.T.; Claasen, J.-H.; Bonde, S.; Kokaia, Z.; Lindvall, O. Inflammation is detrimental for neurogenesis in adult brain. Proc. Natl. Acad. Sci. USA 2003, 100, 13632–13637.
- Yirmiya, R.; Goshen, I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav. Immun. 2011, 25.
- Perez-Dominguez, M.; Ávila-Muñoz, E.; Domínguez-Rivas, E.; Zepeda, A. The detrimental effects of lipopolysaccharide-induced neuroinflammation on adult hippocampal neurogenesis depend on the duration of the pro-inflammatory response. Neural Regen. Res. 2019, 14, 817.
- Monje, M.L. Inflammatory Blockade Restores Adult Hippocampal Neurogenesis. Science 2003, 302, 1760–1765.
- Depino, A.M. Early prenatal exposure to LPS results in anxiety- and depression-related behaviors in adulthood. Neuroscience 2015, 299.
- Sapolsky, R.M.; Meaney, M.J.; McEwen, B.S. The development of the glucocorticoid receptor system in the rat limbic brain. III. Negative-feedback regulation. Dev. Brain Res. 1985, 18, 169–173.
- Videbech, P.; Ravnkilde, B. Hippocampal volume and depression: A meta-analysis of MRI studies. Am. J. Psychiatry 2004, 161, 1957–1966.
- Cobb, J.A.; Simpson, J.; Mahajan, G.J.; Overholser, J.C.; Jurjus, G.J.; Dieter, L.; Herbst, N.; May, W.; Rajkowska, G.; Stockmeier, C.A. Hippocampal volume and total cell numbers in major depressive disorder. J. Psychiatr. Res. 2013, 47, 299–306.
- Koch, S.B.J.; van Ast, V.A.; Kaldewaij, R.; Hashemi, M.M.; Zhang, W.; Klumpers, F.; Roelofs, K. Larger dentate gyrus volume as predisposing resilience factor for the development of trauma-related symptoms. Neuropsychopharmacology 2021.
- Krishnan, V.; Han, M.H.; Graham, D.L.; Berton, O.; Renthal, W.; Russo, S.J.; LaPlant, Q.; Graham, A.; Lutter, M.; Lagace, D.C.; et al. Molecular Adaptations Underlying Susceptibility and Resistance to Social Defeat in Brain Reward Regions. Cell 2007, 131, 391–404.
- Tse, Y.C.; Montoya, I.; Wong, A.S.; Mathieu, A.; Lissemore, J.; Lagace, D.C.; Wong, T.P. A longitudinal study of stress-induced hippocampal volume changes in mice that are susceptible or resilient to chronic social defeat. Hippocampus 2014, 24, 1120–1128.
- Watanabe, Y.; Gould, E.; McEwen, B.S. Stress induces atrophy of apical dendrites of hippocampal CA3 pyramidal neurons. Brain Res. 1992, 588, 341–345.
- Schoenfeld, T.J.; McCausland, H.C.; Morris, H.D.; Padmanaban, V.; Cameron, H.A. Stress and Loss of Adult Neurogenesis Differentially Reduce Hippocampal Volume. Biol. Psychiatry 2017, 82, 914–923.
- Boku, S.; Nakagawa, S.; Toda, H.; Hishimoto, A. Neural basis of major depressive disorder: Beyond monoamine hypothesis. Comput. Graph. Forum 2018, 37, 3–12.
- Schloesser, R.J.; Jimenez, D.V.; Hardy, N.F.; Paredes, D.; Catlow, B.J.; Manji, H.K.; Mckay, R.D.; Martinowich, K. Atrophy of pyramidal neurons and increased stress-induced glutamate levels in CA3 following chronic suppression of adult neurogenesis. Brain Struct. Funct. 2014, 219, 1139–1148.
- Boldrini, M.; Hen, R.; Underwood, M.D.; Rosoklija, G.B.; Dwork, A.J.; Mann, J.J.; Arango, V. Hippocampal angiogenesis and progenitor cell proliferation are increased with antidepressant use in major depression. Biol. Psychiatry 2012, 72, 562–571.
- Boldrini, M.; Santiago, A.N.; Hen, R.; Dwork, A.J.; Rosoklija, G.B.; Tamir, H.; Arango, V.; John Mann, J. Hippocampal granule neuron number and dentate gyrus volume in antidepressant-treated and untreated major depression. Neuropsychopharmacology 2013, 38, 1068–1077.
- Boldrini, M.; Underwood, M.D.; Hen, R.; Rosoklija, G.B.; Dwork, A.J.; John Mann, J.; Arango, V. Antidepressants increase neural progenitor cells in the human hippocampus. Neuropsychopharmacology 2009, 34, 2376–2389.
- Klein, D.N.; Arnow, B.A.; Barkin, J.L.; Dowling, F.; Kocsis, J.H.; Leon, A.C.; Manber, R.; Rothbaum, B.O.; Trivedi, M.H.; Wisniewski, S.R. Early adversity in chronic depression: Clinical correlates and response to pharmacotherapy. Depress. Anxiety 2009, 26, 701–710.
- Boldrini, M.; Galfalvy, H.; Dwork, A.J.; Rosoklija, G.B.; Trencevska-Ivanovska, I.; Pavlovski, G.; Hen, R.; Arango, V.; Mann, J.J. Resilience Is Associated with Larger Dentate Gyrus, While Suicide Decedents with Major Depressive Disorder Have Fewer Granule Neurons. Biol. Psychiatry 2019, 85, 850–862.
- Hamon, M.; Blier, P. Monoamine neurocircuitry in depression and strategies for new treatments. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2013, 45, 54–63.
- Mahar, I.; Bambico, F.R.; Mechawar, N.; Nobrega, J.N. Stress, serotonin, and hippocampal neurogenesis in relation to depression and antidepressant effects. Neurosci. Biobehav. Rev. 2014, 38, 173–192.
- Gao, L.; Gao, T.; Zeng, T.; Huang, P.; Wong, N.K.; Dong, Z.; Li, Y.; Deng, G.; Wu, Z.; Lv, Z. Blockade of Indoleamine 2, 3-dioxygenase 1 ameliorates hippocampal neurogenesis and BOLD-fMRI signals in chronic stress precipitated depression. Aging 2021, 13, 5875–5891.
- Alenina, N.; Klempin, F. The role of serotonin in adult hippocampal neurogenesis. Behav. Brain Res. 2015, 277, 49–57.
- Kulkarni, V.A.; Jha, S.; Vaidya, V.A. Depletion of norepinephrine decreases the proliferation, but does not influence the survival and differentiation, of granule cell progenitors in the adult rat hippocampus. Eur. J. Neurosci. 2002, 16, 2008–2012.
- Masuda, T.; Nakagawa, S.; Boku, S.; Nishikawa, H.; Takamura, N.; Kato, A.; Inoue, T.; Koyama, T. Noradrenaline increases neural precursor cells derived from adult rat dentate gyrus through beta2 receptor. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2012, 36, 44–51.
- Jhaveri, D.J.; Mackay, E.W.; Hamlin, A.S.; Marathe, S.V.; Nandam, L.S.; Vaidya, V.A.; Bartlett, P.F. Norepinephrine directly activates adult hippocampal precursors via β3-adrenergic receptors. J. Neurosci. 2010, 30, 2795–2806.
- Santarelli, L.; Saxe, M.; Gross, C.; Surget, A.; Battaglia, F.; Dulawa, S.; Weisstaub, N.; Lee, J.; Duman, R.; Arancio, O.; et al. Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 2003, 301, 805–809.
- Mateus-Pinheiro, A.; Pinto, L.; Bessa, J.M.; Morais, M.; Alves, N.D.; Monteiro, S.; Patrício, P.; Almeida, O.F.; Sousa, N. Sustained remission from depressive-like behavior depends on hippocampal neurogenesis. Transl. Psychiatry 2013, 3, e210.
- Jedynak, P.; Kos, T.; Sandi, C.; Kaczmarek, L.; Filipkowski, R.K. Mice with ablated adult brain neurogenesis are not impaired in antidepressant response to chronic fluoxetine. J. Psychiatr. Res. 2014, 56, 106–111.
- David, D.J.; Samuels, B.A.; Rainer, Q.; Wang, J.W.; Marsteller, D.; Mendez, I.; Drew, M.; Craig, D.A.; Guiard, B.P.; Guilloux, J.P.; et al. Neurogenesis-Dependent and -Independent Effects of Fluoxetine in an Animal Model of Anxiety/Depression. Neuron 2009, 62, 479–493.
- Bessa, J.M.; Ferreira, D.; Melo, I.; Marques, F.; Cerqueira, J.J.; Palha, J.A.; Almeida, O.F.X.; Sousa, N. The mood-improving actions of antidepressants do not depend on neurogenesis but are associated with neuronal remodeling. Mol. Psychiatry 2009, 14, 764–773.
- Pandarakalam, J.P. Challenges of treatment-resistant depression. Psychiatr. Danub. 2018, 30, 273–284.
- Yamada, J.; Jinno, S. Potential link between antidepressant-like effects of ketamine and promotion of adult neurogenesis in the ventral hippocampus of mice. Neuropharmacology 2019, 158, 107710.
- Geddes, J.R.; Carney, S.M.; Davies, C.; Furukawa, T.A.; Kupfer, D.J.; Frank, E.; Goodwin, G.M. Relapse prevention with antidepressant drug treatment in depressive disorders: A systematic review. Lancet 2003, 361, 653–661.
- Rotheneichner, P.; Lange, S.; O’Sullivan, A.; Marschallinger, J.; Zaunmair, P.; Geretsegger, C.; Aigner, L.; Couillard-Despres, S. Hippocampal neurogenesis and antidepressive therapy: Shocking relations. Neural Plast. 2014, 2014, 723915.
- Nuninga, J.O.; Mandl, R.C.W.; Boks, M.P.; Bakker, S.; Somers, M.; Heringa, S.M.; Nieuwdorp, W.; Hoogduin, H.; Kahn, R.S.; Luijten, P.; et al. Volume increase in the dentate gyrus after electroconvulsive therapy in depressed patients as measured with 7T. Mol. Psychiatry 2020, 25, 1559–1568.
- Gbyl, K.; Rostrup, E.; Raghava, J.M.; Andersen, C.; Rosenberg, R.; Larsson, H.B.W.; Videbech, P. Volume of hippocampal subregions and clinical improvement following electroconvulsive therapy in patients with depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2021, 104.
- Joshi, S.H.; Espinoza, R.T.; Pirnia, T.; Shi, J.; Wang, Y.; Ayers, B.; Leaver, A.; Woods, R.P.; Narr, K.L. Structural plasticity of the hippocampus and amygdala induced by electroconvulsive therapy in major depression. Biol. Psychiatry 2016, 79, 282–292.
- Schloesser, R.J.; Orvoen, S.; Jimenez, D.V.; Hardy, N.F.; Maynard, K.R.; Sukumar, M.; Manji, H.K.; Gardier, A.M.; David, D.J.; Martinowich, K. Antidepressant-like Effects of Electroconvulsive Seizures Require Adult Neurogenesis in a Neuroendocrine Model of Depression. Brain Stimul. 2015, 8, 862–867.
- Jonckheere, J.; Deloulme, J.C.; Dall’Igna, G.; Chauliac, N.; Pelluet, A.; Nguon, A.S.; Lentini, C.; Brocard, J.; Denarier, E.; Brugière, S.; et al. Short- and long-term efficacy of electroconvulsive stimulation in animal models of depression: The essential role of neuronal survival. Brain Stimul. 2018, 11, 1336–1347.
- Petrik, D.; Lagace, D.C.; Eisch, A.J. The neurogenesis hypothesis of affective and anxiety disorders: Are we mistaking the scaffolding for the building? Neuropharmacology 2012, 62, 21–34.
- Tsai, C.Y.; Tsai, C.Y.; Arnold, S.J.; Huang, G.J. Ablation of hippocampal neurogenesis in mice impairs the response to stress during the dark cycle. Nat. Commun. 2015, 6.
- Otte, C.; Gold, S.M.; Penninx, B.W.; Pariante, C.M.; Etkin, A.; Fava, M.; Mohr, D.C.; Schatzberg, A.F. Major depressive disorder. Nat. Rev. Dis. Prim. 2016, 2, 1–20.
- Hodes, G.E.; Kana, V.; Menard, C.; Merad, M.; Russo, S.J. Neuroimmune mechanisms of depression. Nat. Neurosci. 2015, 18, 1386–1393.
- Mazza, M.G.; Lucchi, S.; Tringali, A.G.M.; Rossetti, A.; Botti, E.R.; Clerici, M. Neutrophil/lymphocyte ratio and platelet/lymphocyte ratio in mood disorders: A meta-analysis. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2018, 84.
- Hasselmann, H.; Gamradt, S.; Taenzer, A.; Nowacki, J.; Zain, R.; Patas, K.; Ramien, C.; Paul, F.; Wingenfeld, K.; Piber, D.; et al. Pro-inflammatory Monocyte Phenotype and Cell-Specific Steroid Signaling Alterations in Unmedicated Patients with Major Depressive Disorder. Front. Immunol. 2018, 9.
- Grosse, L.; Hoogenboezem, T.; Ambrée, O.; Bellingrath, S.; Jörgens, S.; de Wit, H.J.; Wijkhuijs, A.M.; Arolt, V.; Drexhage, H.A. Deficiencies of the T and natural killer cell system in major depressive disorder. Brain Behav. Immun. 2016, 54.
- Petralia, M.C.; Mazzon, E.; Fagone, P.; Basile, M.S.; Lenzo, V.; Quattropani, M.C.; Di Nuovo, S.; Bendtzen, K.; Nicoletti, F. The cytokine network in the pathogenesis of major depressive disorder. Close to translation? Autoimmun. Rev. 2020, 19.
- Kim, T.D.; Lee, S.; Yoon, S. Inflammation in Post-Traumatic Stress Disorder (PTSD): A Review of Potential Correlates of PTSD with a Neurological Perspective. Antioxidants 2020, 9, 107.
- Hodes, G.E.; Pfau, M.L.; Leboeuf, M.; Golden, S.A.; Christoffel, D.J.; Bregman, D.; Rebusi, N.; Heshmati, M.; Aleyasin, H.; Warren, B.L.; et al. Individual differences in the peripheral immune system promote resilience versus susceptibility to social stress. Proc. Natl. Acad. Sci. USA 2014, 111, 16136–16141.
- Li, S.; Wang, C.; Wang, W.; Dong, H.; Hou, P.; Tang, Y. Chronic mild stress impairs cognition in mice: From brain homeostasis to behavior. Life Sci. 2008, 82.
- Miller, A.H.; Raison, C.L. The role of inflammation in depression: From evolutionary imperative to modern treatment target. Nat. Rev. Immunol. 2016, 16, 22–34.
- Gill, H.; El-Halabi, S.; Majeed, A.; Gill, B.; Lui, L.M.W.; Mansur, R.B.; Lipsitz, O.; Rodrigues, N.B.; Phan, L.; Chen-Li, D.; et al. The Association between Adverse Childhood Experiences and Inflammation in Patients with Major Depressive Disorder: A Systematic Review. J. Affect. Disord. 2020, 272, 1–7.
- Khandaker, G.M.; Pearson, R.M.; Zammit, S.; Lewis, G.; Jones, P.B. Association of Serum Interleukin 6 and C-Reactive Protein in Childhood with Depression and Psychosis in Young Adult Life. JAMA Psychiatry 2014, 71, 1121.
- Benros, M.E.; Waltoft, B.L.; Nordentoft, M.; Østergaard, S.D.; Eaton, W.W.; Krogh, J.; Mortensen, P.B. Autoimmune Diseases and Severe Infections as Risk Factors for Mood Disorders. JAMA Psychiatry 2013, 70, 812.
- Mousa, A.; Bakhiet, M. Role of Cytokine Signaling during Nervous System Development. Int. J. Mol. Sci. 2013, 14, 3931.
- Cain, D.W.; Cidlowski, J.A. Immune regulation by glucocorticoids. Nat. Rev. Immunol. 2017, 17, 233–247.
- Carlessi, A.S.; Borba, L.A.; Zugno, A.I.; Quevedo, J.; Réus, G.Z. Gut microbiota–brain axis in depression: The role of neuroinflammation. Eur. J. Neurosci. 2021, 53, 222–235.
- Cooper, C.; Moon, H.Y.; Van Praag, H. On the run for hippocampal plasticity. Cold Spring Harb. Perspect. Med. 2018, 8.
- Kempermann, G. Environmental enrichment, new neurons and the neurobiology of individuality. Nat. Rev. Neurosci. 2019, 20, 235–245.
- Voss, M.W.; Soto, C.; Yoo, S.; Sodoma, M.; Vivar, C.; van Praag, H. Exercise and Hippocampal Memory Systems. Trends Cogn. Sci. 2019, 23, 318–333.e6.
- Van Praag, H.; Christie, B.R.; Sejnowski, T.J.; Gage, F.H. Running enhances neurogenesis, learning, and long-term potentiation in mice. Proc. Natl. Acad. Sci. USA 1999, 96, 13427–13431.
- Van Praag, H.; Kempermann, G.; Gage, F.H. Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat. Neurosci. 1999, 2, 266–270.
- Voss, M.W.; Vivar, C.; Kramer, A.F.; van Praag, H. Bridging animal and human models of exercise-induced brain plasticity. Trends Cogn. Sci. 2013, 17, 525–544.
- Kronenberg, G.; Bick-Sander, A.; Bunk, E.; Wolf, C.; Ehninger, D.; Kempermann, G. Physical exercise prevents age-related decline in precursor cell activity in the mouse dentate gyrus. Neurobiol. Aging 2006, 27, 1505–1513.
- Zhao, C.; Teng, E.M.; Summers, R.G.; Ming, G.L.; Gage, F.H. Distinct morphological stages of dentate granule neuron maturation in the adult mouse hippocampus. J. Neurosci. 2006, 26, 3–11.
- van Praag, H.; Schinder, A.F.; Christie, B.R.; Toni, N.; Palmer, T.D.; Gage, F.H. Functional neurogenesis in the adult hippocampus. Nature 2002, 415, 1030–1034.
- Clelland, C.D.; Choi, M.; Romberg, C.; Clemenson, G.D.; Fragniere, A.; Tyers, P.; Jessberger, S.; Saksida, L.M.; Barker, R.A.; Gage, F.H.; et al. A Functional Role for Adult Hippocampal Neurogenesis in Spatial Pattern Separation. Sciences 2009, 325.
- Kobilo, T.; Liu, Q.-R.; Gandhi, K.; Mughal, M.; Shaham, Y.; van Praag, H. Running is the neurogenic and neurotrophic stimulus in environmental enrichment. Learn. Mem. 2011, 18.
- Mustroph, M.L.; Chen, S.; Desai, S.C.; Cay, E.B.; DeYoung, E.K.; Rhodes, J.S. Aerobic exercise is the critical variable in an enriched environment that increases hippocampal neurogenesis and water maze learning in male C57BL/6J mice. Neuroscience 2012, 219.
- Ehninger, D.; Kempermann, G. Regional effects of wheel running and environmental enrichment on cell genesis and microglia proliferation in the adult murine neocortex. Cereb. Cortex 2003, 13, 845–851.
- Tapia-Rojas, C.; Aranguiz, F.; Varela-Nallar, L.; Inestrosa, N.C. Voluntary Running Attenuates Memory Loss, Decreases Neuropathological Changes and Induces Neurogenesis in a Mouse Model of Alzheimer’s Disease. Brain Pathol. 2016, 26.
- Rendeiro, C.; Rhodes, J.S. A new perspective of the hippocampus in the origin of exercise–brain interactions. Brain Struct. Funct. 2018, 223, 2527–2545.
- Piomelli, D. The molecular logic of endocannabinoid signalling. Nat. Rev. Neurosci. 2003, 4, 873–884.
- Ligresti, A.; De Petrocellis, L.; Di Marzo, V. From phytocannabinoids to cannabinoid receptors and endocannabinoids: Pleiotropic physiological and pathological roles through complex pharmacology. Physiol. Rev. 2016, 96, 1593–1659.
- Lutz, B. Neurobiology of cannabinoid receptor signaling. Dialogues Clin. Neurosci. 2020, 22, 207–222.
- Oddi, S.; Scipioni, L.; Maccarrone, M. Endocannabinoid system and adult neurogenesis: A focused review. Curr. Opin. Pharmacol. 2020, 50, 25–32.
- MacCarrone, M.; Guzmán, M.; MacKie, K.; Doherty, P.; Harkany, T. Programming of neural cells by (endo)cannabinoids: From physiological rules to emerging therapies. Nat. Rev. Neurosci. 2014, 15, 786–801.
- Zhang, Z.; Wang, W.; Zhong, P.; Liu, S.J.; Long, J.Z.; Zhao, L.; Gao, H.Q.; Cravatt, B.F.; Liu, Q.S. Blockade of 2-arachidonoylglycerol hydrolysis produces antidepressant-like effects and enhances adult hippocampal neurogenesis and synaptic plasticity. Hippocampus 2015, 25, 16–26.
- Zhong, P.; Wang, W.; Pan, B.; Liu, X.; Zhang, Z.; Long, J.Z.; Zhang, H.T.; Cravatt, B.F.; Liu, Q.S. Monoacylglycerol lipase inhibition blocks chronic stress-induced depressive-like behaviors via activation of mTOR signaling. Neuropsychopharmacology 2014, 39, 1763–1776.
- Jernigan, C.S.; Goswami, D.B.; Austin, M.C.; Iyo, A.H.; Chandran, A.; Stockmeier, C.A.; Karolewicz, B. The mTOR signaling pathway in the prefrontal cortex is compromised in major depressive disorder. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2011, 35, 1774–1779.
- Li, N.; Lee, B.; Liu, R.J.; Banasr, M.; Dwyer, J.M.; Iwata, M.; Li, X.Y.; Aghajanian, G.; Duman, R.S. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 2010, 329, 959–964.
- Paliouras, G.N.; Hamilton, L.K.; Aumont, A.; Joppé, S.E.; Barnabé-Heider, F.; Fernandes, K.J.L. Mammalian target of rapamycin signaling is a key regulator of the transit-amplifying progenitor pool in the adult and aging forebrain. J. Neurosci. 2012, 32, 15012–15026.
- Licausi, F.; Hartman, N.W. Role of mTOR complexes in neurogenesis. Int. J. Mol. Sci. 2018, 19, 1544.
- Chevalier, G.; Siopi, E.; Guenin-Macé, L.; Pascal, M.; Laval, T.; Rifflet, A.; Boneca, I.G.; Demangel, C.; Colsch, B.; Pruvost, A.; et al. Effect of gut microbiota on depressive-like behaviors in mice is mediated by the endocannabinoid system. Nat. Commun. 2020, 11.
- Hill, M.N.; Titterness, A.K.; Morrish, A.C.; Carrier, E.J.; Lee, T.T.Y.; Gil-Mohapel, J.; Gorzalka, B.B.; Hillard, C.J.; Christie, B.R. Endogenous cannabinoid signaling is required for voluntary exercise-induced enhancement of progenitor cell proliferation in the hippocampus. Hippocampus 2010, 20, 513–523.
- Dubreucq, S.; Koehl, M.; Abrous, D.N.; Marsicano, G.; Chaouloff, F. CB1 receptor deficiency decreases wheel-running activity: Consequences on emotional behaviours and hippocampal neurogenesis. Exp. Neurol. 2010, 224, 106–113.
- Gottmann, K.; Mittmann, T.; Lessmann, V. BDNF signaling in the formation, maturation and plasticity of glutamatergic and GABAergic synapses. Exp. Brain Res. 2009, 199, 203–234.
- Foltran, R.B.; Diaz, S.L. BDNF isoforms: A round trip ticket between neurogenesis and serotonin? J. Neurochem. 2016, 138, 204–221.
- Leschik, J.; Eckenstaler, R.; Nieweg, K.; Lichtenecker, P.; Nieweg, K.; Brigadski, T.; Gottmann, K.; Lessmann, V.; Lutz, B. Embryonic stem cells stably expressing BDNF-GFP exhibit a BDNF-release-dependent enhancement of neuronal differentiation. J. Cell Sci. 2013, 126, 5062–5073.
- Numakawa, T.; Odaka, H.; Adachi, N. Actions of brain-derived neurotrophin factor in the neurogenesis and neuronal function, and its involvement in the pathophysiology of brain diseases. Int. J. Mol. Sci. 2018, 19, 3650.
- Edelmann, E.; Leßmann, V.; Brigadski, T. Pre- and postsynaptic twists in BDNF secretion and action in synaptic plasticity. Neuropharmacology 2014, 76, 610–627.
- Duman, R.S.; Monteggia, L.M. A Neurotrophic Model for Stress-Related Mood Disorders. Biol. Psychiatry 2006, 59, 1116–1127.
- Park, H.; Poo, M.M. Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 2013, 14, 7–23.
- Brigadski, T.; Leßmann, V. The physiology of regulated BDNF release. Cell Tissue Res. 2020, 382, 15–45.
- Leßmann, V.; Brigadski, T. Mechanisms, locations, and kinetics of synaptic BDNF secretion: An update. Neurosci. Res. 2009, 65, 11–22.
- Leschik, J.; Eckenstaler, R.; Endres, T.; Munsch, T.; Edelmann, E.; Richter, K.; Kobler, O.; Fischer, K.D.; Zuschratter, W.; Brigadski, T.; et al. Prominent Postsynaptic and Dendritic Exocytosis of Endogenous BDNF Vesicles in BDNF-GFP Knock-in Mice. Mol. Neurobiol. 2019, 56, 6833–6855.
- Groves, N.; O’Keeffe, I.; Lee, W.; Toft, A.; Blackmore, D.; Bandhavkar, S.; Coulson, E.J.; Bartlett, P.F.; Jhaveri, D.J. Blockade of TrkB but not p75NTR activates a subpopulation of quiescent neural precursor cells and enhances neurogenesis in the adult mouse hippocampus. Dev. Neurobiol. 2019, 79, 868–879.
- Donovan, M.H.; Yamaguchi, M.; Eisch, A.J. Dynamic expression of TrkB receptor protein on proliferating and maturing cells in the adult mouse dentate gyrus. Hippocampus 2008, 18, 435–439.
- Kojima, M.; Mizui, T. BDNF Propeptide: A Novel Modulator of Synaptic Plasticity. In Vitamins and Hormones; Academic Press Inc.: Cambridge, MA, USA, 2017; Volume 104, pp. 19–28.
- Tejeda, G.S.; Díaz-Guerra, M. Integral characterization of defective BDNF/TrkB signalling in neurological and psychiatric disorders leads the way to new therapies. Int. J. Mol. Sci. 2017, 18, 268.
- Numakawa, T.; Suzuki, S.; Kumamaru, E.; Adachi, N.; Richards, M.; Kunugi, H. BDNF function and intracellular signaling in neurons. Histol. Histopathol. 2010, 25, 237–258.
- Sasi, M.; Vignoli, B.; Canossa, M.; Blum, R. Neurobiology of local and intercellular BDNF signaling. Pflug. Arch. 2017, 469, 593–610.
- Polyakova, M.; Beyer, F.; Mueller, K.; Sander, C.; Witte, V.; Lampe, L.; Rodrigues, F.; Riedel-Heller, S.; Kratzsch, J.; Hoffmann, K.T.; et al. Serum BDNF levels correlate with regional cortical thickness in minor depression: A pilot study. Sci. Rep. 2020, 10, 14524.
- Karege, F.; Perret, G.; Bondolfi, G.; Schwald, M.; Bertschy, G.; Aubry, J.M. Decreased serum brain-derived neurotrophic factor levels in major depressed patients. Psychiatry Res. 2002, 109, 143–148.
- Sen, S.; Duman, R.; Sanacora, G. Serum Brain-Derived Neurotrophic Factor, Depression, and Antidepressant Medications: Meta-Analyses and Implications. Biol. Psychiatry 2008, 64, 527–532.
- Brunoni, A.R.; Lopes, M.; Fregni, F. A systematic review and meta-analysis of clinical studies on major depression and BDNF levels: Implications for the role of neuroplasticity in depression. Int. J. Neuropsychopharmacol. 2008, 11, 1169–1180.
- Sheline, Y.I. Neuroimaging studies of mood disorder effects on the brain. Biol. Psychiatry 2003, 54, 338–352.
- Karege, F.; Schwald, M.; Cisse, M. Postnatal developmental profile of brain-derived neurotrophic factor in rat brain and platelets. Neurosci. Lett. 2002, 328, 261–264.
- Hashimoto, K. Brain-derived neurotrophic factor as a biomarker for mood disorders: An historical overview and future directions. Psychiatry Clin. Neurosci. 2010, 64, 341–357.
- Fuchikami, M.; Morinobu, S.; Segawa, M.; Okamoto, Y.; Yamawaki, S.; Ozaki, N.; Inoue, T.; Kusumi, I.; Koyama, T.; Tsuchiyama, K.; et al. DNA Methylation Profiles of the Brain-Derived Neurotrophic Factor (BDNF) Gene as a Potent Diagnostic Biomarker in Major Depression. PLoS ONE 2011, 6, e23881.
- Pittenger, C.; Duman, R.S. Stress, depression, and neuroplasticity: A convergence of mechanisms. Neuropsychopharmacology 2008, 33, 88–109.
- Franklin, T.B.; Perrot-Sinal, T.S. Sex and ovarian steroids modulate brain-derived neurotrophic factor (BDNF) protein levels in rat hippocampus under stressful and non-stressful conditions. Psychoneuroendocrinology 2006, 31, 38–48.
- McEwen, B.S. Stress and hippocampal plasticity. Annu. Rev. Neurosci. 1999, 22, 105–122.
- Lucassen, P.J.; Naninck, E.F.G.; van Goudoever, J.B.; Fitzsimons, C.; Joels, M.; Korosi, A. Perinatal programming of adult hippocampal structure and function; Emerging roles of stress, nutrition and epigenetics. Trends Neurosci. 2013, 36, 621–631.
- Arnold, S.E.; Trojanowski, J.Q. Human fetal hippocampal development: I. Cytoarchitecture, myeloarchitecture, and neuronal morphologic features. J. Comp. Neurol. 1996, 367, 274–292.
- Altman, J.; Bayer, S.A. Migration and distribution of two populations of hippocampal granule cell precursors during the perinatal and postnatal periods. J. Comp. Neurol. 1990, 301.
- Brydges, N.M.; Moon, A.; Rule, L.; Watkin, H.; Thomas, K.L.; Hall, J. Sex specific effects of pre-pubertal stress on hippocampal neurogenesis and behaviour. Transl. Psychiatry 2018, 8.
- Naninck, E.F.G.; Hoeijmakers, L.; Kakava-Georgiadou, N.; Meesters, A.; Lazic, S.E.; Lucassen, P.J.; Korosi, A. Chronic early life stress alters developmental and adult neurogenesis and impairs cognitive function in mice. Hippocampus 2015, 25, 309–328.
- Loi, M.; Koricka, S.; Lucassen, P.J.; Joëls, M. Age- and sex-dependent effects of early life stress on hippocampal neurogenesis. Front. Endocrinol. 2014, 5.
- Floriou-Servou, A.; von Ziegler, L.; Waag, R.; Schläppi, C.; Germain, P.-L.; Bohacek, J. The Acute Stress Response in the Multiomic Era. Biol. Psychiatry 2021.
- Gjerstad, J.K.; Lightman, S.L.; Spiga, F. Role of glucocorticoid negative feedback in the regulation of HPA axis pulsatility. Stress 2018, 21.
More