Abnormalities of ion channels and synaptic machineries can cause a variety of neurological diseases. Mutations in genes coding for ion channels cause paroxysmal neurological diseases, such as epilepsy and episodic ataxia. Autoantibodies can also target ion channels and related proteins as well as synaptic machinery proteins to cause limbic encephalitis and immune-mediated cerebellar ataxias. In this brief review, we outline the physiological and molecular features of ion channels and receptors, and summarize the clinical profiles of certain channelopathies and synaptopathies. Following this background, we address two pathophysiological issues; 1) How do ion channel or synapse abnormalities lead to clinical neurological features?, and 2) Why are ion channel and synapse abnormalities limited to a specific brain area? We provide some clues to these fundamental questions.
- ion channels
- synapses
- epilepsy
- episodic ataxia
- limbic encephalitis
- immune-mediated cerebellar ataxias
- Introduction
1. Introduction
Ion channels and synaptic machineries are functional elements underlying elaborate and region-specific functions in the central nervous system (CNS). These functional elements can be the targets in certain pathological conditions, paroxysmal diseases and autoimmune diseases. In the former etiology, deficits in genes of ion channels elicit paroxysmal neurological features, mainly epilepsy [1-3][1][2][3], episodic ataxias (EAs) [4-7] [4][5][6][7] and migraine. In some etiology autoantibodies toward ion channels and receptors may play a particular role [8]. Historically, myasthenia gravis and Lambert-Eaton syndrome are well-known forms of this type of autoimmune diseases, in both of which neuromuscular junctions are blocked by anti-acetylcholine receptor and anti-voltage gated calcium channel (VGCC), respectively [8]. Recent evidence also indicates that autoantibodies could be involved in some limbic encephalitis [9-13] [9][10][11][12][13] and cerebellar ataxias (CAs) [14-22][14][15][16][17][18][19][20][21][22].
Herein, we provide a brief review of the physiological features of ion channels and synaptic machineries, mainly receptors, and summarize clinical profiles of these channelopathies and synaptopathies. Based on these translational approaches, the current review focuses on two pathophysiological issues; 1) how do impairments in ion channels or synapses lead to neurological symptoms?, and 2) why are ion channels and synapse in a specific area disturbed? Challenges to these issues will yield key insights that lead to deeper mechanistic understanding of channelopathies and synaptopathies.
2. Overview of ion channels and synaptic machineries
- Overview of ion channels and synaptic machineries
2.1. General features of ion channels and receptors
Action potential, arriving at the axonal terminal, opens the voltage-gated Ca-channel. The increase in intracellular Ca concentration ([Ca2+]in) at the nanodomain of the presynaptic active zone induces neurotransmitter release through Ca-dependent exocytotic molecular machinery. Then, the diffused neurotransmitter binds and activates agonist-driven receptor-ion channels at the postsynaptic membrane. Some neurotransmitters/modulators activate ion channel through the G-protein coupled receptors. The electrical integration of excitatory postsynaptic potentials (EPSPs) and inhibitory postsynaptic potentials (IPSCs) determines whether the postsynaptic neuron generates an action potential or not.
Voltage-gated ion-channel: The voltage-gated ion channel is functionally composed of an ion selective filter, voltage-dependent gate and a pore region. Certain types of ion-channels possess voltage-gated inactivation mechanism. The ion-selective filter can permit particular ions, such as K+, Na+, Ca2+ or Cl-, according to their molecular structure. With regard to the structure of the K+ ion selective filter, four subunits surround the central pore, and each subunit has a characteristic amino acids sequence, Thr-Val-Gly-Tyr-Gly, which is called as “K+ channel signature sequence”. These amino acids construct a molecular structure to dehydrate hydrated K+, and the dehydrated K+ can pass through the filter, in an exclusive manner. The voltage-dependent gate has a “voltage sensor region” composed by charged amino acids. In general, the structural alternation of “open-close” obeys thermodynamics in a stochastic manner. When the membrane potential depolarizes, the "open" channel probability of the voltage-dependent gate of K+ channels increases based on the Bolzmann distribution. In the pore region, the ions movement is passively driven by their own electrochemical gradient across the membrane.
Ion channels and exocytic machinery at the presynaptic terminal
The arrival of the action potential at the presynaptic terminal activates the voltage-gated Ca-
channels, such as P/Q-type and/or N-type Ca2+-channel. This is followed by an increase in Ca2+-influx flow through the opened Ca2+-channel. With [Ca2+]in near the Ca2+-channel (nanodomain) around 50 mM. Ca2+ binds to synaptotagmin, the main Ca2+ sensor of Ca2+-dependent neurotransmitter release, which stops the synaptotagmin from impeding the fusion of the synaptic vesicle membrane to the plasma membrane. This is followed by neurotransmitters release into the synaptic cleft via exocytosis. Exocytosis is mediated by binding of SNARE core proteins, i.e., VAMP, a vesicular membrane protein, SNAP-25 and syntaxin, plasma membrane proteins. Dissociation of the bound SNARE-core complex requires ATP decomposition by NSF. An ATPase enzyme. Vesicular membrane, fused into the plasma membrane is recovered via AP-2, clathrin-dependent endocytosis, and recycled as synaptic vesicle membrane. Autoreceptors expressed at the presynaptic terminal suppress neurotransmitter release via a negative feedback loop.
Receptors at the postsynaptic terminal
At the postsynaptic membrane, the receptors for neurotransmitters are localized by the synaptic scaffold proteins. Synaptic adhesion molecules also contribute to the localization of the postsynaptic membrane. At the excitatory synapse, activation of the transmitter-gated ion channels, such as glutamate, nicotinic-ACh- and 5-HT3 receptors elicits opening of the cation channel, leading to the depolarization of membrane potential. When the membrane potential reaches the threshold voltage, an action potential is generated in the postsynaptic neuron. Examples of transmitter-gated ion-channels for glutamate receptors include the AMPA-type, NMDA-type and KA-type. At the inhibitory synapse, GABA or Gly activates GABAA-receptor or Gly-receptor, respectively, and cause hyperpolarization via opening of Cl- -channel. G-protein-coupled receptors (GPCRs) are also expressed in the postsynaptic cell. Representative receptors coupled with Gas are b-Adrenergic, dopamine-receptor type 1 (DA1) and 5-HT4. Receptors coupled with Gai/o are GABAB, D2, 5-HT1 while receptors coupled with Gaq/11 are mGluR1 and 5-HT2.
Table 1. Representative ion channels and receptors in the CNS neuron.
|
|
Subtype |
Ion selectivity/ Coupled Gα |
Distribution |
|
Voltage gated ion channel |
|||
|
Na+ channel |
Nav1 |
Na+ |
axon, soma, dendrite |
|
K+ channel |
Kv1 |
K+ |
axon, soma, dendrite |
|
Kv2 |
K+ |
soma |
|
|
Kv3 |
K+ |
presynaptic terminal, soma |
|
|
Kv4 |
K+ |
soma, dendrite |
|
|
KCa1 (BK) |
K+ |
axon, soma, dendrite |
|
|
KCa2 (SK) |
K+ |
axon, dendrite |
|
|
Ca2+ channel |
Cav2.1 (P/Q) |
Ca2+ |
presynaptic terminal, soma, dendrite |
|
Cav2.2 (N) |
Ca2+ |
presynaptic terminal, soma, dendrite |
|
|
Transmitter-gated ion channel |
|||
|
Glutamate |
AMPA-type |
Na+, K+,(Ca2+) |
postsynaptic membrane |
|
NMDA-type |
Na+, K+, Ca2+ |
postsynaptic membrane |
|
|
KA-type |
Na+, K+, |
postsynaptic membrane |
|
|
ACh |
nicotinic |
Na+, K+, Ca2+ |
postsynaptic membrane |
|
Serotonin |
5-HT3 |
Na+, K+, Ca2+ |
postsynaptic membrane |
|
GABA |
GABAA |
Cl- |
postsynaptic membrane |
|
Gly |
|
Cl- |
postsynaptic membrane |
|
G-protein-coupled receptors |
|||
|
Glutamate |
mGluR1 |
q/11 |
postsynaptic membrane |
|
mGluR2 |
i/o |
presynaptic terminal |
|
|
ACh |
muscalinic M1 |
q/11 |
postsynaptic membrane |
|
muscalinic M2 |
i/o |
postsynaptic membrane |
|
|
Noradrenalin |
α1AR |
q/11 |
presynaptic terminal |
|
βAR |
s |
postsynaptic membrane |
|
|
Dopamine |
D1 |
s |
postsynaptic membrane |
|
D2 |
i/o |
postsynaptic membrane |
|
|
Serotonin |
5-HT1 |
i/o |
postsynaptic membrane |
|
5-HT2 |
q/11 |
presynaptic terminal |
|
|
GABA |
GABAB |
i/o |
postsynaptic membrane |
2.2. Role of Ion Channels and Synaptic Machineries in Cerebellar Specific Functions
2.2. Role of Ion Channels and Synaptic Machineries in Cerebellar Specific Functions
Notably, a cross talk between ion channels and synaptic receptors induces long-term synaptic plasticity in the cerebellum. The roles of ion channels and receptors can be explained also in terms of cerebellar specific functions.
The cerebellar neuronal network is involved in coordinative control of motor and cognitive functions [26-29][23][24][25][26]. Various internal forward and inverse models are assumed to be embedded within the cerebellum [30][27]. For example, using the internal forward model, the cerebellum serves as a predictive controller [31,32][28][29]. In motor coordination, the internal model predicts sensory consequences in response to issued motor commands [32][29]. Two parameters; synergy and timing, are computed using this internal model, which is updated through learning. One representative hypothesis is the Marr-Albus-Ito theory, which postulates that the strength of the parallel fiber (PF)-Purkinje cell (PC) synapse is plastically adjusted via climbing fiber’s (CF’s) error signals [30][27]. These specific functions depend on the properties of the ion channels and synaptic transmission in the cerebellum. The unique features of the Na+ and K+ channels underlie the exact spike firing during timing controls [33-36][30][31][32][33], whereas the characteristic functions of the Ca2+ signaling system and the subsequent synaptic plasticity contribute to the continuous update of the internal model [27,30,37][24][27][34].
Conjunctive stimulation of CF and PF is reported to cause long-term depression (LTD) of PF-PC synaptic transmission, both in vivo [38] [35] and in vitro [39-41][36][37][38]. The CF inputs elicit an increase in [Ca2+]in through VGCC (P/Q-type) (Figure 1) [42][39]. The PF inputs in dendritic spines activate the metabotropic glutamate receptor (mGluR)-PLCβ-IP3 signaling pathways, and such activation elicits Ca2+ release from the Ca2+-stores in the endoplasmic reticulum (ER) through IP3 receptors and, as a result, increases [Ca2+]in (Figure 2) [43][40]. The simultaneous activation of mGluR1 and VGCC (P/Q-type) elicits a series of events, including an increase in [Ca2+]in to levels higher than the additive level [44][41], which leads to the activation of PKCα, which in turn phosphorylates GluA2-C terminus, ultimately leading to the detachment of AMPAR, including phosphorylated GluA2, from the scaffold protein and its internalization with PICK1 in AP2- and clathrin-dependent manners (Figure 1) [37][34]. Gene-manipulations that cause deficits in LTD were found to elicit abnormalities in VOR-adaptation, eye-blink conditioning, and ataxia [45][42].
Synaptic plasticity is maintained by trans-synaptic synaptic adhesion molecules that bidirectionally coordinate synapse formation, restructuring, and elimination. Among the synaptic adhesion molecules in the cerebellar cortex, neurexin is widely expressed as the presynaptic transmembrane protein [46][43]. Furthermore, cerebellin, secreted from PF terminals, mediates the neurexin-cerebellin-glutamate receptor delta (GluRδ) interaction [47][44]. GluRδ is a postsynaptic transmembrane protein localized at the PF-PC synapse. Interestingly, cerebellin gene-deleted mice lack LTD and show ataxic movements [48][45].
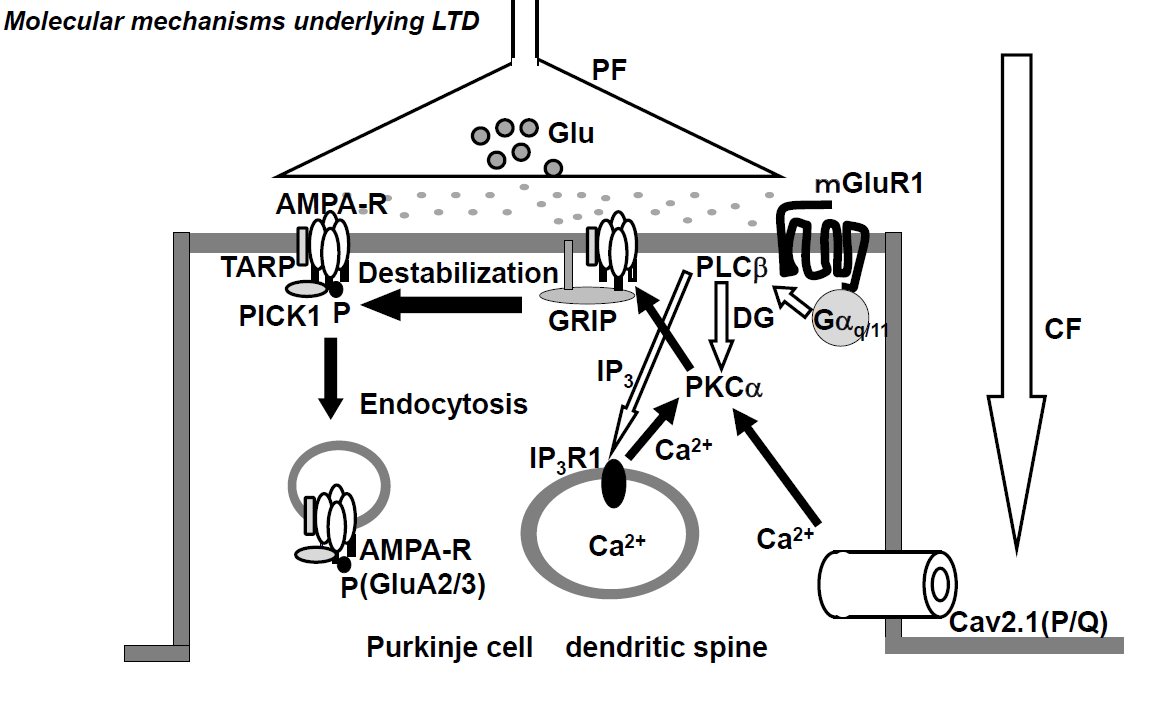
Figure 1. Schematic diagram of long-term depression (LTD) at excitatory synapses between parallel fibers and Purkinje cells. The climbing fiber input elicits complex spikes through the activation of dendritic P/Q type Ca2+ channels, leading to an increase in intracellular calcium concentration ([Ca2+]in). On the other hand, the parallel fiber input activates metabotropic glutamate receptor-PLCβ-IP3 signaling pathways, resulting in an increase in [Ca2+]in. The conjunctive activation of these two pathways increases the [Ca2+]in level more than the additive level. The high [Ca2+]in level activates PKCα, and PKCα phosphorylates GluA2 of the AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptor, which results in detachment of the AMPA receptor from the scaffold proteins and its internalization with PICK1 in AP2- and clathrin-dependent manner.
CF; climbing fiber, PF; parallel fiber, Glu; glutamate; AMAPA-R; AMPA receptor, mGluR1; metabotropic glutamate receptor, Cav2.1 (P/Q); P/Q type Ca2+ voltage-gated channel, PLC; phospholipase C, PKC; protein kinase C, IP3; Inositol triphosphate 3, GRIP; Glutamate receptor interactive protein, TARP; transmembrane AMPA receptor regulatory proteins, PICK1; protein interacting with C kinase.
3. Clinical phenotypes of abnormalities in ion channels and synaptic machineries in the cerebral cortex
- Clinical phenotypes of abnormalities in ion channels and synaptic machineries in the cerebral cortex
53.1. Epilepsy
Table 2 shows mutations in variety of ion channels and GABA receptors responsible for epilepsy [1-3] [1][2][3] Imbalance between excitation and inhibition is assumed to be a key factor underlying epilepsy. Gain-of–function or loss-of function mutations can result in enhancement of action potentials or excitatory synaptic transmission, or impairment of inhibitory neurons, leading to hyperexcitability [3].
53.2. Episodic ataxia
Autosomal-dominant episodic ataxias (EAs), characterized by recurrent episodes of CAs, represent a clinically and genetically heterogeneous group of disorders [4-6][4][5][6]. Ataxic episodes are usually of short duration and often triggered by specific stimuli [4-6][4][5][6]. Seven subtypes have been recognized so far [4, 5][4][5]. Amongst them, the EA types 1 and 2 are the most common (Table 2). EA1 is caused by mutations of the potassium channel gene KCNA1, and evident clinically by myokymias between the episodes of ataxia. On the other hand, EA2 involves mutations in CACNA1A, a P/Q-type VGCC-encoding gene [4-6] [4][5][6] and the patients often show interictal downbeat nystagmus with other cerebellar ocular dysfunctions, together with slowly progressive ataxia and vermian atrophy. Therapeutically, acetazolamide is beneficial in the control of EA2 though it is less effective in EA1 [5, 7][5][7]. 4-aminopyridine may provide benefits.
53.23. Limbic encephalitis
Accumulating evidence has confirmed the association of limbic encephalitis with autoantibodies toward potassium channel-related proteins, glutamate receptors, and GABA receptors [9-13,[9][10][11][12][13][46][47][48][49][50][51][52] 49-55] (Table 3). The clinical features include short-term memory loss, confusion, cognitive deficits, abnormal behavior, and seizures [9-13, 49-55][9][10][11][12][13][46][47][48][49][50][51][52]. CSF examination shows pleocytosis, high protein levels, and oligoclonal bands in some patients [9-13, 49-55][9][10][11][12][13][46][47][48][49][50][51][52]. FLAIR and T2 MRI studies usually show increased signals in the temporal lobe [9-13, 49-55][9][10][11][12][13][46][47][48][49][50][51][52]. Some of these patients have paraneoplastic conditions. The majority of patients received immunotherapy, though the benefits vary according to the subtype or the presence of neoplasm.
53.24. Immune-mediated cerebellar ataxias
Immune-mediated cerebellar ataxias (IMCAs) include diverse etiologies in which autoimmune mechanisms insult the cerebellum [14-22][14][15][16][17][18][19][20][21][22]. In most of the cases IMCAs are associated with neuronal death (particularly PCs), probably by activation of cellular immunity [56][53]. However, in few rarer cases, etiologies are considered to involve autoantibodies-induced functional impairments of ion channels and synaptic transmissions. Table 3 shows the association of CAs with anti-ion channel or their related proteins antibodies (Abs) and anti-synaptic machinery proteins Abs [9-13, 49-55, 57-61]. Notably, the frequency of CAs is classified into four patterns (1) common phenotype (anti-VGCC, mGluR1, GAD65 Abs), (2) sometimes associated (anti-Caspr2, DPPX, GluRδ Abs), (3) occasionally associated (anti-AMPAR and GlycineR Abs), and (4) rarely associated (anti-LgI1, GABA shows the association of CAs with anti-ion channel or their related proteins antibodies (Abs) and anti-synaptic machinery proteins Abs [9][10][11][12][13][46][47][48][49][50][51][52][54][55][56][57][58]. Notably, the frequency of CAs is classified into four patterns (1) common phenotype (anti-VGCC, mGluR1, GAD65 Abs), (2) sometimes associated (anti-Caspr2, DPPX, GluRδ Abs), (3) occasionally associated (anti-AMPAR and GlycineR Abs), and (4) rarely associated (anti-LgI1, GABAAR, and GABABR Abs) [62].R Abs) [59].
Table 2. Inherited mutations of ion channels responsible for epilepsy and episodic ataxia.
CACNA1A |
(Cav(2).1) |
|
Condition |
Impaired channel |
Gene (channel subunit) |
|
Epilepsy |
Sodium channel |
SCN1A(α subunit of Nav1.1), SCN1B(β1), SCN2A(α subunit of Nav1.1) |
|
Potassium channel |
KCNQ2(Kv7.2), KCNQ3(Kv7.3), KCNMA1(BK), KCNA1(Kv1.1), KCNC3(Kv3.3) |
|
|
Calcium channel |
CACNA1H(α1H subunit of Cav3.2) |
|
|
GABAAR |
GABRA1(α1), GABRB3(β3), GABRG2(γ2) |
|
|
Episodic ataxia |
Potassium channel |
KCNA1(Kv1.1) |
|
Calcium channel |
CACNA1A(Cav(2).1) |
Table 3. Autoantibodies associated with autoimmune limbic encephalitis and immune-mediated cerebellar ataxias.
|
Condition |
Frequency |
Autoantibodies |
|
Autoimmune limbic encephalopathy |
Common phenotype |
Anti-LgI1, Caspr2, DPPX Abs, Anti-NMDAR, AMPAR, GABAAR, ABABR, |
|
Immune-mediated cerebellar ataxias |
Common phenotype |
Anti-VGCC, mGluR1, GAD65 Abs |
|
Sometimes associated |
Anti-Caspr2, DPPX, GluRδ Abs |
|
|
Occasionally associated |
Anti-AMPAR, GlycineR Abs |
|
|
Rarely associated |
Anti-LgI1, GABAAR, GABABR Abs |
|
Condition |
Impaired channel |
Gene (channel subunit) |
|
Epilepsy |
Sodium channel |
SCN1A(α subunit of Nav1.1), SCN1B(β1), SCN2A(α subunit of Nav1.1) |
|
Potassium channel |
KCNQ2(Kv7.2), KCNQ3(Kv7.3), KCNMA1(BK), KCNA1(Kv1.1), KCNC3(Kv3.3) |
|
|
Calcium channel |
CACNA1H(α1H subunit of Cav3.2) |
|
|
GABAAR |
GABRA1(α1), GABRB3(β3), GABRG2(γ2) |
|
|
Episodic ataxia |
Potassium channel |
KCNA1(Kv1.1) |
|
Calcium channel |
LgI1: Leucine-rich glioma-inactivated 1; Caspr2: Contactin-associated protein-like 2; DPPX: Dipeptidyl-peptidase-like protein-6; VGCC: P/Q-type voltage-gated calcium channel; mGluR1: metabotropic glutamate receptor 1; GluRδ: glutamate receptor delta; GAD: glutamate acid decarboxylase
- Conclusions
4. Conclusions
64.1. How do ion channel or synapse abnormalities lead to clinical neurological features?
In all cases, ion channel and synapses abnormalities are associated with imbalance between neural excitation and inhibition, resulting in impaired formation of neural signals in the local neural circuits. Especially, epilepsy is likely to occur when such imbalance elicits hyperexcitability in the cerebral cortex. In addition to this general mechanism, more specific mechanisms can operate in the cerebellum. For example, mutations in the VGCC-encoding gene CACNA1A and the presence of anti-VGCC and anti-mGluR Abs often impair the characteristic functions of the Ca2+ signaling system with subsequent internalization of the AMPA receptors. In other words, the signal cascades underlying synaptic plasticity (LTD) are final common pathways in these channelopathies and synaptopathies. Thus, key molecular apparatuses for region-specific functions are preferentially targeted.
64.2. Why are ion channel and synapse abnormalities limited to a specific brain area?
Inherited ion channel mutations can affect the entire CNS. Autoantibodies to ion channels and synaptic machinery proteins have potential actions over the CNS. Thus, additional factors should be taken into consideration. One possible explanation is that neural excitability is tightly regulated and that alterations in some ion channels or synapses can be compensated for by the activities of other ion channels and/or synaptic alterations [1]. Such abilities for compensation might differ depending the brain region.
Alternatively, in autoimmune diseases, the cerebellar region-specific vulnerability might be due to local inflammation [62][59]. That is, despite the wide distribution of AMPAR and GABAR in the cerebellum, the association of CAs with autoantibodies toward these receptors is rare. In these etiologies, limbic encephalitis is the main clinical phenotype. A breakdown of the blood-brain barrier (BBB) is necessary for infiltration of effector T and B cells [8], and when such breakdown occurs, these lymphocytes overexpress adhesion molecules, which enhances their passage from the circulation to the parenchyma. Importantly, the permeability of BBB is different between the hippocampus and the cerebellum [63][60]. Recent studies have shown that the CSF is locally produced and absorbed between capillaries and the interstitial fluid [64][61], which implies that although CSF autoantibodies are humoral factors, their actions can be local. Taken together, neurons, which are involved in local inflammation, are the main targets of autoantibodies whose actions are local.
We hope that this unifying issue will enhance our understanding of the pathological roles of ion channels and synaptic machineries in neurological diseases.
References
- Kullmann, D.M.; Waxman, S.G. Neurological channelopathies: New insights into disease mechanisms and ion channel function. Physiol. 2010, 588, 18223-1827.
- Kullmann, D.M. Neurological channelopathies. Annu. Neurosci. 2010, 33, 151-172.
- Catterall, W.A.; Dib-Hajj, S.; Meisler, M.H.; Pietrobon, D. Inherited neuronal ion channelopathies: New windows on complex neurological diseases. Neurosci. 2008, 28, 11768-11777.
- Kipfer, S.; Strupp, M. The clinical spectrum of autosomal-dominant episodic ataxias. Disord. Clin. Pract. 2014, 1, 245-290.
- Baloh RW. Episodic ataxias 1 and 2. Clin. Neurol. 2012, 103, 595-602.
- Jen, J.C.; Wan, J. Episodic ataxias. Clin. Neurol. 2018, 148, 521-529.
- Strupp, M.; Thurtell, M.J.; Shaikh, A.G.; Brandt, T.; Zee, D.S.; Leigh, R.J. Pharmacotherapy of vestibular and ocular motor disorders, including nystagmus. Neurol.2011, 258, 1207–1222.
- Mitoma, M.; Manto, M. Neuroimmune Diseases; From Cells to the Living Brain; Springer Nature: Basel, Switzerland, 2019.
- Lancaster, E.; Dalmau, J. Neuronal autoantigens-pathogenesis, associated disorders and antibody testing. Rev. Neurol. 2012, 8, 380–392.
- Graus, F.; Saiz, A.; Dalmau, J. Antibodies and neuronal autoimmune disorders of the J. Neurol. 2010, 257, 509–517.
- Lancaster, E.; Martinex-Hernandez, E.; Dalmau, J. Encephalitis and antibodies to synaptic and neuronal cell surface proteins. Neurology 2011, 77, 179–189.
- Dalmau, J.; Rosenfeld, M.R. Autoimmune encephalitis update. Oncol. 2014, 16, 771–778.
- Dalmau, J.; Geis, C.; Graus, F. Autoantibodies to synaptic receptors and neuronal cell surface protein in autoimmune diseases of the central nervous system. Rev. 2017, 97, 839–887.
- Hadjivassiliou, M. Immune-mediated acquired ataxias. Clin. Neurol. 2012, 103, 189–199.
- Mitoma, H.; Adhikari, K.; Aeschlimann, D.; Chattopadhyay, P.; Hadjivassiliou, M.; Hampe, C.S.; Honnorat, J.; Joubert, B.; Kakei, S.; Lee, J.; et al. Consensus Paper: Neuroimmune mechanisms of cerebellar ataxias. Cerebellum 2016, 15, 2313–2332.
- Mitoma, H.; Hadjivassiliou, M.; Honnorat, J. Guidelines for treatment of immune-mediated cerebellar ataxias. Cerebellum Ataxias 2015, 2, 14.
- Mitoma, H.; Manto, M.; Hampe, C.S. Immune-mediated cerebellar ataxias: Practical guidelines and therapeutic challenges. Neuropharmacol. 2018, doi:10.2174/1570159X16666180917105033.
- Joubert, B.; Rotásky, J.; Honnorat, J. Nonparaneoplastic autoimmune cerebellar ataxia. Clin. Neurol. 2018, 155, 313–332.
- Joubert, B.; Honnorat, J. Nonparaneoplastic autoimmune cerebellar ataxia. Opin. Neurol. 2019, 32, 484–492.
- MacKinonn, R. Potassium channels. FEBS Lett. 2003, 555, 62-65.
- Hille, B. Ion channels of excitable membranes: Third edition. Sinauer, Sunderland, Massachusetts, U.S.A. 2001.
- Schuneggenburger, R.; Rosenmund, C. Molecular mechanisms governing Ca2+ regulation of evoked and spontaneous release. Neurosci. 2015, 18, 935-941.
- Südhof, T.C. Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron 2013, 80, 675-690.
- Scheefhals, N.; MacGillavry, H.D. Functional organization of postsynaptic glutamate receptors. Cell Neurosci. 2018, 91, 82-94.
- Manglik, A.; Kobilka, B. The role of protein dynamics in GPCR function: insights from the 2AR and rhodopsin. Opin. Cell Biol. 2014, 27, 136-143.
- Eccles, J.C.; Ito, M.; Szentagothai, J. The Cerebellum as A Neuronal Machine; Springer: New York, NY, USA, 1967.
- Ito, M. The Cerebellum and Neural Control; Raven Press: New York, NY, USA, 1984.
- Manto, M.; Bower, J.M.; Conforto, A.B.; Delgado-García, J.M.; da Guarda, S.N.; Gerwig, M.; Habas, C.; Hagura, N.; Ivry, R.B.; Mariën, P.; et al. Consensus Paper: Roles of the cerebellum in motor control—The diversity of ideas on cerebellar involvement in movement. Cerebellum 2012, 11, 457–847.
- Schmahmann, J.D.; Caplan, D. Cognitive, emotion and the cerebellum. Brain 2006, 129, 290–292.
- Ito, M. Cerebellar long-term depression: Characterization, signal transduction, and functional roles. Rev. 2001, 81, 1143–1195.
- Popa, L.S.; Hewitt, A.L.; Ebner, T.J. Predictive and feedback performance errors are signaled in the simple spike discharge of individual Purkinje cells. Neurosci. 2012, 32, 15345–15358.
- Tanaka, H.; Ishikawa, T.; Lee, J.; Kakei, S..The cerebro-cerebellum as a locus of forward model: A review. Syst. Neurosci. 2020, 14, 19.
- Afshari, F.S.; Ptak, K.; Khaliq, Z.M.; Grieco, T.M.; Slater, N.T.; McCrimmon, D.R.; Raman, I.M. Resurgent Na currents in four classes of neurons of the cerebellum. Neurophysiol. 2004, 92, 2831–4283.
- Masoli, S.; Solinas, S.; D’Angelo, E. Action potential processing in a detailed Purkinje cell model reveals a critical role for axonal compartmentalization. Cell Neurosci. 2015, 9, 47.
- McKay, B.E.; Molineux, M.L.; Mehaffey, W.H.; Turner, R.W. Kv1 K+ channels control Purkinje cell output to facilitate postsynaptic rebound discharge in deep cerebellar neurons. Neurosci. 2005, 25, 1481–1492.
- Zagha, E.; Lang, E.J.; Rudy, B. Kv3.3 channels at the Purkinje cell soma are necessary for generation of the classical complex spike waveform. Neurosci. 2008, 28, 1291–300.
- Wang, Y.T.; Linden, D.J. Expression of cerebellar long-term depression requires postsynaptic clathrin-mediated endocytosis. Neuron 2000, 25, 635–647.
- Ito, M.; Sakurai, M.; Tongroach, P. Climbing fibre induced depression of both mossy fibre responsiveness and glutamate sensitivity of cerebellar Purkinje cells. Physiol. 1982, 324, 113–134.
- Hirano, T.; Ohmori, H. Voltage-gated and synaptic currents in rat Purkinje cells in dissociated cell cultures. Natl. Acad. Sci. USA 1986, 83, 1945–1949.
- Sakurai, M. Synaptic modification of parallel fibre-Purkinje cell transmission in in vitro guinea-pig cerebellar slices. Physiol. 1987, 394, 463–480.
- Linden, D.J.; Connor, J.A. Participation of postsynaptic PKC in cerebellar long-term depression in culture. Science 1991, 254, 1656–1659.
- Linás, R.; Sugimori, M. Electrophysiological properties of in vitro Purkinje cell dendrites in mammalian cerebellar slices. Physiol. 1980, 305, 197–213.
- Ito, M. The molecular organization of cerebellar long-term depression. Nature Rev. Neurosci. 2002, 3, 896-902.
- Wang, S.S.; Denk, W.; Hausser, M. Coincidence detection in single dendritic spines mediated by calcium release. Neurosci. 2000, 3, 1266–1273.
- Ito, M.; Yamaguchi, K.; Nagao, S.; Yamazaki, T. Long-term depression as a model of cerebellar plasticity. Brain Res. 2014, 210, 1–30.
- Yuzaki, M. Two classes of secreted synaptic organizers in the central nervous system. Rev. Physiol. 2018, 80, 243–262.
- Matsuda, K.; Miura, E.; Miyazaki, T.; Kakegawa, W.; Emi, K.; Narumi, S.; Fukazawa, Y.; Ito-Ishida, A.; Kondo, T.; Shigemoto, R.; et al. Cbln1 is a ligand for an orphan glutamate receptor delta 2, a bidirectional synapse organizer. Science 2010, 328, 363–368.
- Ito-Ishida, A.; Okabe, S.; Yuzaki, M. The role of Cbln1 on Purkinje cell synapse formation. Res. 2014, 83, 64–68.
- Navarro, V.; Kas, A.; Apartis, E.; Chami, L.; Rogemond, V.; Levy, P.; Psimaras, D.; Habert, M.O.; Baulac, M.; Delattre, J.Y.; et al. Motor cortex and hippocampus are the two main cortical targets in LGI1-antibody encephalitis. Brain 2016, 139, 1079–1093.
- Joubert, B.; Gobert, F.; Thomas, L.; Saint-Martin, M.; Desestret, V.; Convers, P.; Rogemond, V.; Picard, G.; Ducray, F.; Psimaras, D.; et al. Autoimmune episodic ataxia with anti-CASPR2 antibody-associated encephalitis. Neuroimmunol. Neuroinflamm. 2017, 14, e371.
- Tobin, W.O.; Lennon, V.A.; Komorowski, L.; Probst, C.; Clardy, S.L.; Aksamit, A.J.; Lucchinetti, C.F.; Matsumoto, J.Y.; Pittock, S.J.; Sandroni, P.; et al. DPPX potassium channel antibody; frequency, clinical accompaniments and outcomes in 20 patients. Neurology 2014, 83, 1797–1803.
- Dalmau, J.; Gleichman, A.J.; Hughes, E.G.; Rossi, J.E.; Peng, X.; Lai, M.; Dessain, S.K.; Rosenfeld, M.R.; Balice-Gordon, R.; Lynch, D.R. Anti-NMDA-receptor encephalitis: Case series and analysis of the effect of antibodies. Lancet Neurol. 2008, 7, 1091–1098.
- Höftberger, R.; van Sonderen, A.; Leypoldt, F.; Houghton, D.; Geschwind, M.; Gelfand, J.; Paredes, M.; Sabater, L.; Saiz, A.; Titulaer, M.J.; Graus, F.; et al. Encephalitis and AMPA receptor antibodies. Neurology 2015, 84, 2403–2412.
- Spatola, M.; Petit-Pedrol, M.; Simabukuro, M.M.; Armangue, T.; Castro, F.J.; Barcelo Artigues, M.I.; Julià Benique, M.R.; Benson, L.; Gorman, M.; Felipe, A.; et al. Investigations in GABAA receptor antibody-associated encephalitis. Neurology 2017, 88, 1012–1020.
- Jarius, S.; Steinmeyer, F.; Knobel, A.; Streitberger, K.; Hotter, B.; Horn, S.; Heuer, H.; Schreiber, S.J.; Wilhelm, T.; Trefzer, U.; et al. GABAB receptor antibodies in paraneoplastic cerebellar ataxia. Neuroimmunol. 2013, 256, 94–96.
- Venkatraman, A.; Opal P. Paraneoplastic cerebellar degeneration with anti-Yo antibodies-a review. Clin. Transl.l Neurol. 2016, 3, 655-663.
- Graus, F.; Lang, B.; Pozo-Rosich, P.; Saiz, A.; Casamitjana, R.; Vincent, A. P/Q type calcium-channel antibodies in paraneoplastic cerebellar degeneration with lung cancer. Neurology 2002, 59, 764–766.
- Sillevis Smitt, P.; Kinoshita, A.; De Leeuw, B.; Moll, W.; Coesmans, M.; Jaarsma, D.; Henzen-Logmans, S.; Vecht, C.; De Zeeuw, C.; Sekiyama, N.; et al. Paraneoplastic cerebellar ataxia due to antibodies toward against a glutamate receptor. Engl. J. Med. 2000, 342, 21–27.
- Mitoma, H.; Manto, M.; Hampe, C.S. Pathogenic roles of glutamate decarboxylase 65 autoantibodies in cerebellar ataxias. Immunol. Res. 2017, 2913297.
- Hirai, H.; Launey, T.; Mikawa, S.; Torashima, T.; Yanagihara, D.; Kasaura, T.; Miyamoto, A.; Yuzaki, M. New role of delta2-glutamate receptors in AMPA receptor trafficking and cerebellar function. Neurosci. 2003, 6, 869–876.
- Carvajal-González, A.; Leite, M.I.; Waters, P.; Woodhall, M.; Coutinho, E.; Balint, B.; Lang, B.; Pettingill, P.; Carr, A.; Sheerin, U.M.; et al. Glycine receptor antibodies in PERM and related syndromes: Characteristics, clinical features and outcomes. Brain 2014, 137(pt8), 2178–2192.
- Mitoma, H.; Honnorat, J.; Ymaguchi, K.; Manto, M. Fundamental mechanisms of autoantibody-induced impairments on ion channels and synapses in immune-mediated cerebellar ataxias. J. Mol. Sci. 2020, 21, 4936.
- Villaseñor, R.; Kuennecke, B.; Ozmen, L.; Ammann, M.; Kugler, C.; Grüninger, F.; Loetscher, H.; Freskgård, P.O.; Collin, L. Region-specific permeability of the blood-brain barrier upon pericyte loss. L. Blood Flow Metab. 2017, 37, 3683–3694.
- Orešković, D.; Klarica, M. A new look at cerebrospinal fluid circulation. Fluids Barriers CNS 2014, 11, 16.
References
- Kullmann, D.M.; Waxman, S.G. Neurological channelopathies: New insights into disease mechanisms and ion channel function. Physiol. 2010, 588, 18223-1827.
- Kullmann, D.M. Neurological channelopathies. Annu. Neurosci. 2010, 33, 151-172.
- Catterall, W.A.; Dib-Hajj, S.; Meisler, M.H.; Pietrobon, D. Inherited neuronal ion channelopathies: New windows on complex neurological diseases. Neurosci. 2008, 28, 11768-11777.
- Kipfer, S.; Strupp, M. The clinical spectrum of autosomal-dominant episodic ataxias. Disord. Clin. Pract. 2014, 1, 245-290.
- Baloh RW. Episodic ataxias 1 and 2. Clin. Neurol. 2012, 103, 595-602.
- Jen, J.C.; Wan, J. Episodic ataxias. Clin. Neurol. 2018, 148, 521-529.
- Strupp, M.; Thurtell, M.J.; Shaikh, A.G.; Brandt, T.; Zee, D.S.; Leigh, R.J. Pharmacotherapy of vestibular and ocular motor disorders, including nystagmus. Neurol.2011, 258, 1207–1222.
- Mitoma, M.; Manto, M. Neuroimmune Diseases; From Cells to the Living Brain; Springer Nature: Basel, Switzerland, 2019.
- Lancaster, E.; Dalmau, J. Neuronal autoantigens-pathogenesis, associated disorders and antibody testing. Rev. Neurol. 2012, 8, 380–392.
- Graus, F.; Saiz, A.; Dalmau, J. Antibodies and neuronal autoimmune disorders of the J. Neurol. 2010, 257, 509–517.
- Lancaster, E.; Martinex-Hernandez, E.; Dalmau, J. Encephalitis and antibodies to synaptic and neuronal cell surface proteins. Neurology 2011, 77, 179–189.
- Dalmau, J.; Rosenfeld, M.R. Autoimmune encephalitis update. Oncol. 2014, 16, 771–778.
- Dalmau, J.; Geis, C.; Graus, F. Autoantibodies to synaptic receptors and neuronal cell surface protein in autoimmune diseases of the central nervous system. Rev. 2017, 97, 839–887.
- Hadjivassiliou, M. Immune-mediated acquired ataxias. Clin. Neurol. 2012, 103, 189–199.
- Mitoma, H.; Adhikari, K.; Aeschlimann, D.; Chattopadhyay, P.; Hadjivassiliou, M.; Hampe, C.S.; Honnorat, J.; Joubert, B.; Kakei, S.; Lee, J.; et al. Consensus Paper: Neuroimmune mechanisms of cerebellar ataxias. Cerebellum 2016, 15, 2313–2332.
- Mitoma, H.; Hadjivassiliou, M.; Honnorat, J. Guidelines for treatment of immune-mediated cerebellar ataxias. Cerebellum Ataxias 2015, 2, 14.
- Mitoma, H.; Manto, M.; Hampe, C.S. Immune-mediated cerebellar ataxias: Practical guidelines and therapeutic challenges. Neuropharmacol. 2018, doi:10.2174/1570159X16666180917105033.
- Joubert, B.; Rotásky, J.; Honnorat, J. Nonparaneoplastic autoimmune cerebellar ataxia. Clin. Neurol. 2018, 155, 313–332.
- Joubert, B.; Honnorat, J. Nonparaneoplastic autoimmune cerebellar ataxia. Opin. Neurol. 2019, 32, 484–492.
- MacKinonn, R. Potassium channels. FEBS Lett. 2003, 555, 62-65.
- Hille, B. Ion channels of excitable membranes: Third edition. Sinauer, Sunderland, Massachusetts, U.S.A. 2001.
- Schuneggenburger, R.; Rosenmund, C. Molecular mechanisms governing Ca2+ regulation of evoked and spontaneous release. Neurosci. 2015, 18, 935-941.
- Eccles, J.C.; Ito, M.; Szentagothai, J. The Cerebellum as A Neuronal Machine; Springer: New York, NY, USA, 1967.
- Ito, M. The Cerebellum and Neural Control; Raven Press: New York, NY, USA, 1984.
- Manto, M.; Bower, J.M.; Conforto, A.B.; Delgado-García, J.M.; da Guarda, S.N.; Gerwig, M.; Habas, C.; Hagura, N.; Ivry, R.B.; Mariën, P.; et al. Consensus Paper: Roles of the cerebellum in motor control—The diversity of ideas on cerebellar involvement in movement. Cerebellum 2012, 11, 457–847.
- Schmahmann, J.D.; Caplan, D. Cognitive, emotion and the cerebellum. Brain 2006, 129, 290–292.
- Ito, M. Cerebellar long-term depression: Characterization, signal transduction, and functional roles. Rev. 2001, 81, 1143–1195.
- Popa, L.S.; Hewitt, A.L.; Ebner, T.J. Predictive and feedback performance errors are signaled in the simple spike discharge of individual Purkinje cells. Neurosci. 2012, 32, 15345–15358.
- Tanaka, H.; Ishikawa, T.; Lee, J.; Kakei, S..The cerebro-cerebellum as a locus of forward model: A review. Syst. Neurosci. 2020, 14, 19.
- Afshari, F.S.; Ptak, K.; Khaliq, Z.M.; Grieco, T.M.; Slater, N.T.; McCrimmon, D.R.; Raman, I.M. Resurgent Na currents in four classes of neurons of the cerebellum. Neurophysiol. 2004, 92, 2831–4283.
- Masoli, S.; Solinas, S.; D’Angelo, E. Action potential processing in a detailed Purkinje cell model reveals a critical role for axonal compartmentalization. Cell Neurosci. 2015, 9, 47.
- McKay, B.E.; Molineux, M.L.; Mehaffey, W.H.; Turner, R.W. Kv1 K+ channels control Purkinje cell output to facilitate postsynaptic rebound discharge in deep cerebellar neurons. Neurosci. 2005, 25, 1481–1492.
- Zagha, E.; Lang, E.J.; Rudy, B. Kv3.3 channels at the Purkinje cell soma are necessary for generation of the classical complex spike waveform. Neurosci. 2008, 28, 1291–300.
- Wang, Y.T.; Linden, D.J. Expression of cerebellar long-term depression requires postsynaptic clathrin-mediated endocytosis. Neuron 2000, 25, 635–647.
- Ito, M.; Sakurai, M.; Tongroach, P. Climbing fibre induced depression of both mossy fibre responsiveness and glutamate sensitivity of cerebellar Purkinje cells. Physiol. 1982, 324, 113–134.
- Hirano, T.; Ohmori, H. Voltage-gated and synaptic currents in rat Purkinje cells in dissociated cell cultures. Natl. Acad. Sci. USA 1986, 83, 1945–1949.
- Sakurai, M. Synaptic modification of parallel fibre-Purkinje cell transmission in in vitro guinea-pig cerebellar slices. Physiol. 1987, 394, 463–480.
- Linden, D.J.; Connor, J.A. Participation of postsynaptic PKC in cerebellar long-term depression in culture. Science 1991, 254, 1656–1659.
- Linás, R.; Sugimori, M. Electrophysiological properties of in vitro Purkinje cell dendrites in mammalian cerebellar slices. Physiol. 1980, 305, 197–213.
- Ito, M. The molecular organization of cerebellar long-term depression. Nature Rev. Neurosci. 2002, 3, 896-902.
- Wang, S.S.; Denk, W.; Hausser, M. Coincidence detection in single dendritic spines mediated by calcium release. Neurosci. 2000, 3, 1266–1273.
- Ito, M.; Yamaguchi, K.; Nagao, S.; Yamazaki, T. Long-term depression as a model of cerebellar plasticity. Brain Res. 2014, 210, 1–30.
- Yuzaki, M. Two classes of secreted synaptic organizers in the central nervous system. Rev. Physiol. 2018, 80, 243–262.
- Matsuda, K.; Miura, E.; Miyazaki, T.; Kakegawa, W.; Emi, K.; Narumi, S.; Fukazawa, Y.; Ito-Ishida, A.; Kondo, T.; Shigemoto, R.; et al. Cbln1 is a ligand for an orphan glutamate receptor delta 2, a bidirectional synapse organizer. Science 2010, 328, 363–368.
- Ito-Ishida, A.; Okabe, S.; Yuzaki, M. The role of Cbln1 on Purkinje cell synapse formation. Res. 2014, 83, 64–68.
- Navarro, V.; Kas, A.; Apartis, E.; Chami, L.; Rogemond, V.; Levy, P.; Psimaras, D.; Habert, M.O.; Baulac, M.; Delattre, J.Y.; et al. Motor cortex and hippocampus are the two main cortical targets in LGI1-antibody encephalitis. Brain 2016, 139, 1079–1093.
- Joubert, B.; Gobert, F.; Thomas, L.; Saint-Martin, M.; Desestret, V.; Convers, P.; Rogemond, V.; Picard, G.; Ducray, F.; Psimaras, D.; et al. Autoimmune episodic ataxia with anti-CASPR2 antibody-associated encephalitis. Neuroimmunol. Neuroinflamm. 2017, 14, e371.
- Tobin, W.O.; Lennon, V.A.; Komorowski, L.; Probst, C.; Clardy, S.L.; Aksamit, A.J.; Lucchinetti, C.F.; Matsumoto, J.Y.; Pittock, S.J.; Sandroni, P.; et al. DPPX potassium channel antibody; frequency, clinical accompaniments and outcomes in 20 patients. Neurology 2014, 83, 1797–1803.
- Dalmau, J.; Gleichman, A.J.; Hughes, E.G.; Rossi, J.E.; Peng, X.; Lai, M.; Dessain, S.K.; Rosenfeld, M.R.; Balice-Gordon, R.; Lynch, D.R. Anti-NMDA-receptor encephalitis: Case series and analysis of the effect of antibodies. Lancet Neurol. 2008, 7, 1091–1098.
- Höftberger, R.; van Sonderen, A.; Leypoldt, F.; Houghton, D.; Geschwind, M.; Gelfand, J.; Paredes, M.; Sabater, L.; Saiz, A.; Titulaer, M.J.; Graus, F.; et al. Encephalitis and AMPA receptor antibodies. Neurology 2015, 84, 2403–2412.
- Spatola, M.; Petit-Pedrol, M.; Simabukuro, M.M.; Armangue, T.; Castro, F.J.; Barcelo Artigues, M.I.; Julià Benique, M.R.; Benson, L.; Gorman, M.; Felipe, A.; et al. Investigations in GABAA receptor antibody-associated encephalitis. Neurology 2017, 88, 1012–1020.
- Jarius, S.; Steinmeyer, F.; Knobel, A.; Streitberger, K.; Hotter, B.; Horn, S.; Heuer, H.; Schreiber, S.J.; Wilhelm, T.; Trefzer, U.; et al. GABAB receptor antibodies in paraneoplastic cerebellar ataxia. Neuroimmunol. 2013, 256, 94–96.
- Venkatraman, A.; Opal P. Paraneoplastic cerebellar degeneration with anti-Yo antibodies-a review. Clin. Transl.l Neurol. 2016, 3, 655-663.
- Graus, F.; Lang, B.; Pozo-Rosich, P.; Saiz, A.; Casamitjana, R.; Vincent, A. P/Q type calcium-channel antibodies in paraneoplastic cerebellar degeneration with lung cancer. Neurology 2002, 59, 764–766.
- Sillevis Smitt, P.; Kinoshita, A.; De Leeuw, B.; Moll, W.; Coesmans, M.; Jaarsma, D.; Henzen-Logmans, S.; Vecht, C.; De Zeeuw, C.; Sekiyama, N.; et al. Paraneoplastic cerebellar ataxia due to antibodies toward against a glutamate receptor. Engl. J. Med. 2000, 342, 21–27.
- Mitoma, H.; Manto, M.; Hampe, C.S. Pathogenic roles of glutamate decarboxylase 65 autoantibodies in cerebellar ataxias. Immunol. Res. 2017, 2913297.
- Hirai, H.; Launey, T.; Mikawa, S.; Torashima, T.; Yanagihara, D.; Kasaura, T.; Miyamoto, A.; Yuzaki, M. New role of delta2-glutamate receptors in AMPA receptor trafficking and cerebellar function. Neurosci. 2003, 6, 869–876.
- Carvajal-González, A.; Leite, M.I.; Waters, P.; Woodhall, M.; Coutinho, E.; Balint, B.; Lang, B.; Pettingill, P.; Carr, A.; Sheerin, U.M.; et al. Glycine receptor antibodies in PERM and related syndromes: Characteristics, clinical features and outcomes. Brain 2014, 137(pt8), 2178–2192.
- Mitoma, H.; Honnorat, J.; Ymaguchi, K.; Manto, M. Fundamental mechanisms of autoantibody-induced impairments on ion channels and synapses in immune-mediated cerebellar ataxias. J. Mol. Sci. 2020, 21, 4936.
- Villaseñor, R.; Kuennecke, B.; Ozmen, L.; Ammann, M.; Kugler, C.; Grüninger, F.; Loetscher, H.; Freskgård, P.O.; Collin, L. Region-specific permeability of the blood-brain barrier upon pericyte loss. L. Blood Flow Metab. 2017, 37, 3683–3694.
- Orešković, D.; Klarica, M. A new look at cerebrospinal fluid circulation. Fluids Barriers CNS 2014, 11, 16.