Ecosystem disturbances and resulting decreases of host species richness have been associated with the emergence of zoonotic diseases. Among vertebrates, rodents are important reservoirs of numerous viruses, including some with significant impacts on public health. Exploring the viral diversity in Neotropical rodents, we provide significant insights into zoonotic viruses in Amazonia, and emphasize that habitats and host’s dietary ecology drive viral diversity. Linking richness and abundance of viruses to the ecology and responses to habitat disturbance of their hosts should be starting points for a better understanding of viral emergence, prediction of at-risk situation, and implementation of early control and mitigation measures.
- virome
- alpha diversity
- rodents
- Amazonia
- viral ecology
12. Introduction
Viruses conquered all living systems, infecting microbes and more complex organisms, such as plants, invertebrates, and vertebrates. Their small genomes with high mutation rates [1] give them the ability to evolve and adapt quickly to new environments and potentially the ability to infect new hosts. The development of metagenomic approaches [2] improved knowledge of the extent of viral diversity and of the host spectra of several viral families [3], led to the discovery of new viral genotypes, helping understand their evolutionary history [4]. But a large number of viral species remain unknown and many biomes continue to be unexplored [5]. In the context of natural habitat disturbances, the disruptions of population dynamics favor contacts between species, cross-species transmissions, spill-over, amplification, spread of viruses, and increased contacts with wild fauna and may lead to the emergence of infectious diseases [6][7][8][6–8], 70% of which originated from animals and mainly from wildlife [9]. Recent examples of virus spill-overs, such as the severe acute respiratory syndrome (SARS) in China, the Middle East respiratory syndrome (MERS) in Saudi Arabia, the Ebola epidemic in West Africa, and more recently the COVID-19 if a zoonotic origin is confirmed, have had severe public health and economic consequences [10][11][12][10-12]. Hence, the identification of potentially zoonotic viruses, and the understanding of their transmission mechanisms, gained attention [13][14][15][16][13-16].
Several groups of vertebrates, such as primates, birds, bats, and rodents, are major virus reservoirs [17].
Rodentia is composed of approximately 2277 species [18], occupying most of the ecosystems, from highly anthropized to pristine natural habitats. This diversity, along with fast-paced lives, population dynamics, opportunism, and synanthropism make them efficient amplifiers, spreaders, and transmitters of viruses [19][20], playing key roles in viral emergence phenomena [21][22][23][24]. Amazonia is known for its high biodiversity of mammals, plants, invertebrates, and microbes [25], suggesting high viral diversity and high number of potential zoonotic viruses [26][27]. But Amazonia also remains one of the least explored areas. The few studies investigating the viral diversity have been conducted with bats only [28][29][30].
is composed of approximately 2277 species [18], occupying most of the ecosystems, from highly anthropized to pristine natural habitats. This diversity, along with fast-paced lives, population dynamics, opportunism, and synanthropism make them efficient amplifiers, spreaders, and transmitters of viruses [19,20], playing key roles in viral emergence phenomena [21-24]. Amazonia is known for its high biodiversity of mammals, plants, invertebrates, and microbes [25], suggesting high viral diversity and high number of potential zoonotic viruses [26-27]. But Amazonia also remains one of the least explored areas. The few studies investigating the viral diversity have been conducted with bats only [28-30].
In French Guiana, Northern Amazonia, 36 rodent species are present, and occur in various types of forest, savannah, agricultural areas, peri-urban zones, and urban zones, and several species have been described to host viruses important for human health [22][31][32][33][34][22,31-34]. Using metagenomics approach, we explored how habitats and species shape the viral diversity, exploring seven Neotropical rodent species of various ecological traits and habitat requirements.
23. Data, Model, Applications and Influences
2.1. Data and Mmodel
2.1.1. Sampling and Llaboratory Pprocedures
Rodents were trapped in Northern French Guiana, with Sherman (Sherman Trap Co., Tallahassee, FL, USA) and BTTm traps (Besançon Trap Service mécanique, Besançon, France) on 12 sites during the 2001–2014 period, in disturbed and pristine forests, savannahs, and peri-urban habitats. Rodents were caught alive, brought back to the laboratory facilities, anesthetized, and sampled for blood and/or euthanized to preserve organs, kidney and spleen. Animals were sampled following welfare guidelines [35], the use of genetic resources was approved by the French Ministry of the Environment (reference ABSCH-IRCC-FR-252439-).
Virome study focused on Proechimys cuvieri and P. guyannensis (Echimyidae), and Zygodontomys brevicauda, Oecomys bicolor, O. auyantepui, Hylaeamys yunganus, and Hylaeamys megacephalus (Cricetidae), for a total of 187 individuals and 442 organs and sera. Prior to processing, samples from the same species, the same organs, and the same environment were pooled, resulting in 36 different pools. All pools were then processed as previously described [36].
High-throughput sequencing was carried out at the genomics center of the Institut Pasteur, Paris. Shotgun libraries were prepared by standard Illumina protocols using 1 µg of total genomic DNA. Each sample was tagged according to its provenance (species, organs, and habitats) using Illumina adaptor-specific primers. High-throughput shotgun metagenomic sequencing was carried using Illumina MiSeq or Illumina HiSeq 2500 platforms.
2.1.2. Bioinformatics Aanalysis
As a pre-analysis step, reads were cleaned using FaQCs [37] to filter out erroneous reads. Then, MEGAHT [38][39][38,39] was used to create contigs by a de bruijn graph based de novo assembly process taking cleaned reads as input (Step 1, Figure 1). BWA-MEM [40] and Samtools [41] were then used in order to obtain the number of reads aligning to each contig (Step 2, Figure 1). Finally, the taxonomic assignment of contigs was performed using a homology search against the nucleotide and protein databases with respectively BLASTn and BLAST x searches [42][43][42,43] (Step 3, Figure 1). These two data sets were filtered to the e-values and coverage according to both BLASTn and BLASTx (e-value = 10−5; coverage ≥ 250 nt or 83 amino acids in length) to consolidate the results. The remaining contigs were used for counting in the results. Bacteriophage sequences, not expected in “organ” viromes were eliminated for diversity analysis. Data were plotted and a heatmap was created using Rstudio, “pheatmap” library. The viral genomes’ completeness of assigned contigs was tested using CHECKV (version v0.7.0) and its associated database [44]. Finally, a matrix corresponding to the number of viral reads at the genus/subfamily level for each species-habitat couple was built for statistical analysis (Step 4, Figure 1).
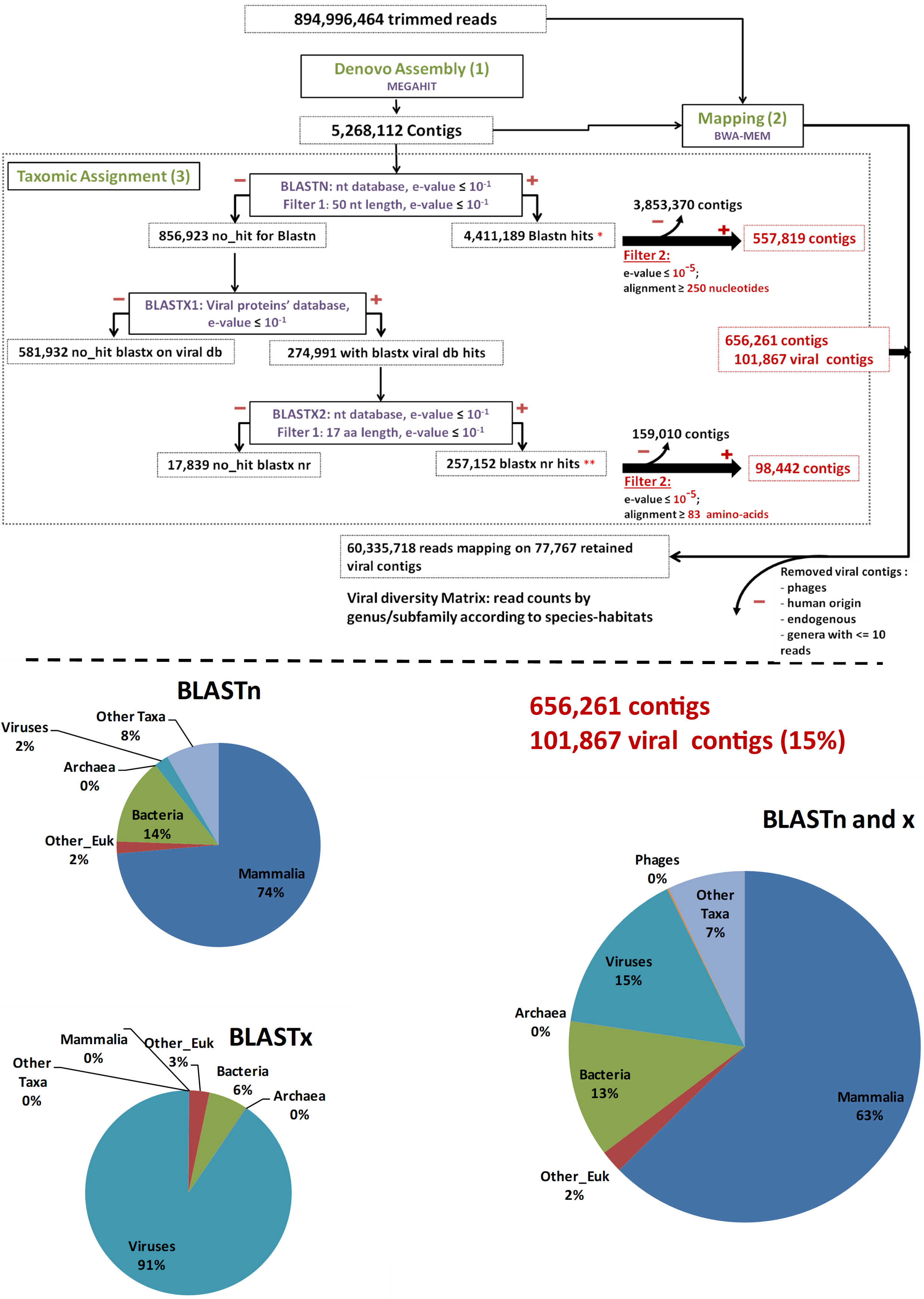
Figure 1. Main bioinformatic processing steps, data streams, and resulting taxonomic categories. “Other_Euk”: Other (non-mammalian eukaryotes).
2.1.3. Analysis of Viral Dviral diversity
To explore how viral diversity is related to habitat type, analyses were run for the four species present in at least two different habitats (pristine and disturbed forest for P. guyannensis, P. cuvieri, and H. megacephalus; disturbed forest, savannah, and peri-urban areas for Z. brevicauda) using the number of mapped reads on contigs assigned to each viral genus or subfamily. We computed diversity indices for each of the nine species and habitat combinations. A synthesis between the most commonly accepted indices has been proposed [45] as a family of indices inspired in statistical physics [46]. The Hill α-diversity index values were generated with α at 0 (= richness), 0.25, 0.5, 0.75, 1 (=Shannon entropy), and 2 (=inverse of Simpson’s dominance index): more α is high, more importance is given to common or dominant species.
Table 1. Richness and Rényi’s entropy values for different values of α between 0 and 2 for the nine species–habitat combinations.
|
|
P.guy.PF |
P.guy.DF |
P.cuv.PF |
P.cuv.DF |
H.meg.PF |
H.meg.DF |
Z.bre.SV |
Z.bre.DF |
Z.bre.PU |
|
Richness |
17 |
16 |
14 |
12 |
13 |
21 |
12 |
11 |
10 |
|
Renyi(α=0) |
2.94 |
2.83 |
2.77 |
2.71 |
2.89 |
3.18 |
3.04 |
2.77 |
2.56 |
|
Renyi(α=0.25) |
2.08 |
1.69 |
1.42 |
1.49 |
1.69 |
1.47 |
1.31 |
1.11 |
0.81 |
|
Renyi(α= 0.5) |
1.55 |
0.97 |
0.94 |
0.89 |
0.87 |
0.65 |
0.70 |
0.49 |
0.16 |
|
Renyi(α=0.75) |
1.21 |
0.59 |
0.81 |
0.67 |
0.48 |
0.37 |
0.51 |
0.29 |
0.04 |
|
Renyi(α=1) |
0.99 |
0.40 |
0.76 |
0.58 |
0.30 |
0.25 |
0.42 |
0.20 |
0.01 |
|
Renyi(α=2) |
0.60 |
0.17 |
0.72 |
0.44 |
0.13 |
0.12 |
0.25 |
0.09 |
0.00 |
2.2. Applications
2.2.1. The virome of Amazonian Rrodents
Overall, 453,865,988 paired-end raw reads (907,731,976 individual reads) were obtained. After trimming by FaQcs, 96.62–99.56% of the reads were kept, totaling 894,996,464 reads. These cleaned reads were assembled de novo for a total of 5,268,112 contigs using MEGAHIT. The number of contigs ranged from 25,215 to 347,957, with a mean number of 146,336 contigs/sample.
After megaBLASTn, 4,411,189 (83.73%) contigs were assigned to an organism, among which 50,431 were attributed to viruses. The remaining 856,923 (16.27%) unassigned contigs were first submitted to a BLASTx search against the in-house viral protein database (BLASTx1) (Figure 1). Only 274,991 contigs matched viral proteins, and they were all put through a second BLASTx step against the entire nr protein database (BLASTx2) (Figure 1). Overall, after this second BLASTx step (BLASTx2), 257,152 contigs were re-assigned to viruses, 3776 were assigned to other types of organisms, and 17,839 were not assigned at all (Figure 1).
To further avoid artifacts and false-positive results, the virus-assigned contigs were filtered at a coverage of ≥250 bp for BLASTn or ≥83 amino acids for BLASTx results and an e-value ≤10−5 (Filter 2), resulting in 101,867 viral contigs, accounting for 1.93% of the total initial number of contigs and 15% of the assigned contigs (Figure 1).
Among the 101,867 viral contigs, 945 assigned to bacteriophages were discarded from further analyses. After removal of contigs with less than 11 reads mapped, 77,767 viral contigs were kept for further analyses. Among them, 118 contigs submitted to CHECKV were high quality, with completeness above 90%, and 72, 23, and 23 contigs were assigned to Anelloviridae, Circoviridae, and Genomoviridae, respectively.
2.2.2. Viral Diversity through Species and Environments
The 77,767 viral contigs obtained were assigned to 27 families known to infect vertebrates, invertebrates, plants, and amoeba (Figure 2). Viral family presence varied largely across species–habitat categories. A pattern of ubiquity was observed for some vertebrate viruses, such as the Genomoviridae. On the other hand, some viruses, especially plant and invertebrate viruses, were restricted to the few species–habitat categories. Six plant-infecting viral families were detected from eight species–habitats. The most common plant-infecting viral family was the Tombusviridae present in five species–habitats, followed by the Luteoviridae, Partitiviridae, and Phycodnaviridae. Five viral families of insect and invertebrate tropism were detected in six different species–habitats, the most common family was Polycipiviridae. Chuviridae, Iflaviridae, Nudiviridae, Picornaviridae were present but far less detected. A total of 11 viral families strictly associated with vertebrates were detected. ssDNA virus families such as Anelloviridae, Circoviridae, Parvoviridae, and Polyomaviridae were detected in 12, nine, eight, and five species–habitats, respectively. Adenoviridae (dsDNA virus) were found only in P. cuvieri from disturbed forest. RNA viruses (Riboviria) accounted for six families. Several positive-sense RNA viral families were also detected: Astroviridae, Arteriviridae, Flaviviridae (Hepacivirus), and Matonaviridae. Matonaviridae were found in only one species–habitat (P. cuvieri in pristine forest), and Arteriviridae were detected only in P. guyannensis from disturbed forest. By contrast, Flaviviridae and Astroviridae had a wider distribution across species–habitats, found in 12 of 12 and nine of 12 species–habitats. Alphavirus (Togaviridae) and Phlebovirus (Phenuiviridae), and the family Rhabdoviridae, all recognized as potential vector-borne viruses, were detected only in P. cuvieri from disturbed forest.
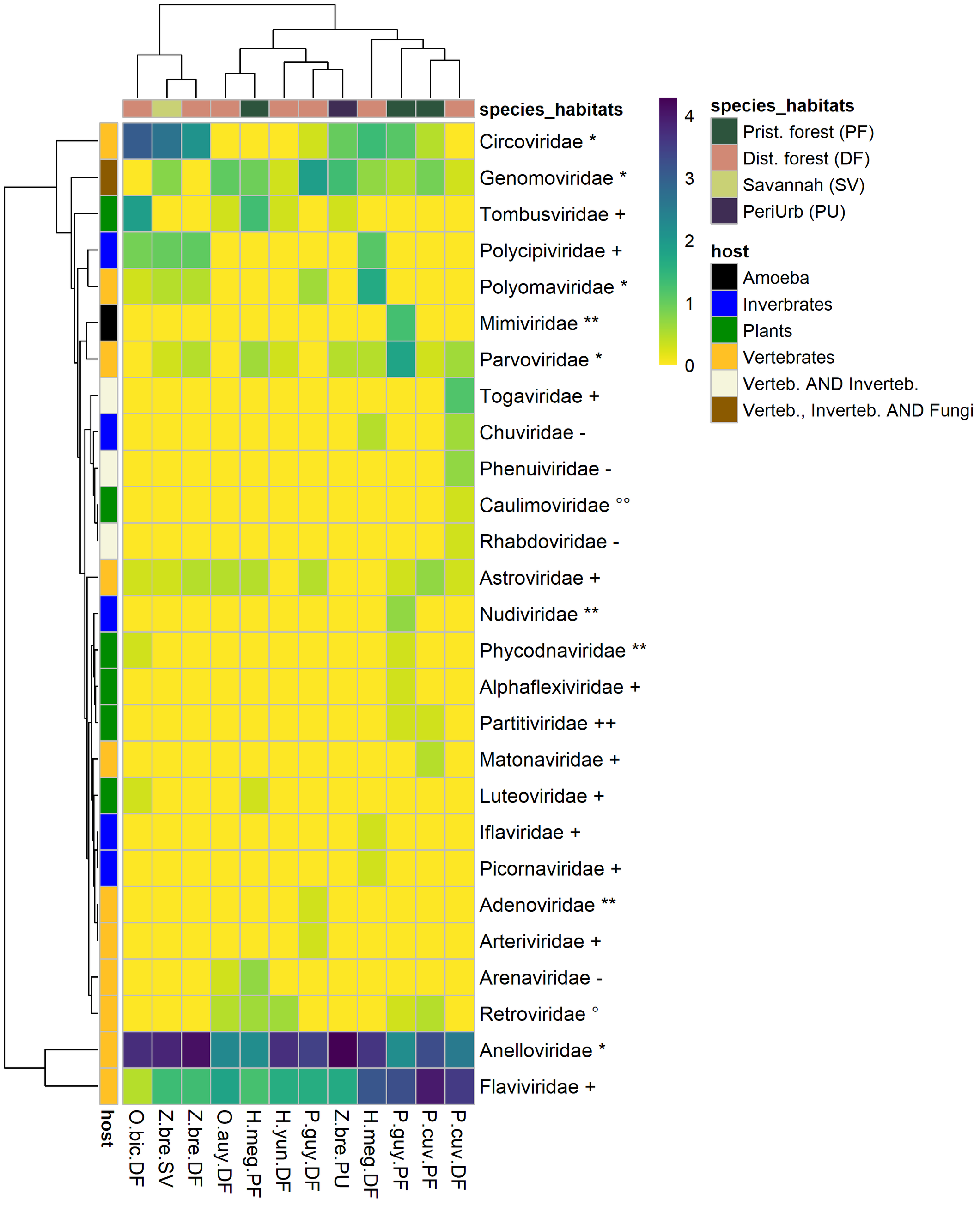
Figure 2. Heatmap of viral families’ numbers of contigs by species–habitat. Each cell representing a viral family in a species-habitat contains log (1 + N), (where N is the number of contigs assigned to a viral family in a species–habitat). The left row represents host-type (vertebrate, invertebrate, plants). Viral family names are marked with genome type, as follows: ** = dsDNA, * = ssDNA, ++ = dsRNA, + = ssRNA (+), - = ssRNA (-), °° = DNA-retrotranscribing, ° = RNA-retrotranscribing. P.guy.PF: Proechimys guyannensis from pristine forest, P.guy.DF: Proechimys guyannensis from disturbed forest, P.cuv.PF: Proechimys cuvieri from pristine forest, P.cuv.DF: Proechimys cuvieri from disturbed forest, H.meg.PF: Hylaeamys megacephalus from pristine forest, H.meg.DF: Hylaeamys megacephalus from pristine forest, Z.bre.SV: Zygodontomys brevicauda from savannah, Z.bre.DF: Zygodontomys brevicauda from disturbed forest, Z.bre.PU: Zygodontomys brevicauda from peri-urban areas.
2.2.3. Viral Diversities
Viral richness ranged from 10 for Zygodontomys in peri-urban habitats and 21 for H. megacephalus in disturbed forests. For P. guyannensis and P. cuvieri, the richness (α = 0) was higher in pristine forest than in disturbed forest. For Z. brevicauda, the diversity was higher in savannahs, followed by disturbed forests, and was lowest in peri-urban habitats. For H. megacephalus, the diversity was higher in disturbed forests for α < 0.25 but higher in pristine forests as soon as α > 0.25, i.e. as soon as more weight is given to common species (Table 1).
2.3. Influences
2.3.1. Lessons for Uundertanding Sshaping of Vviral Ddiversity
Over the past decade, virome studies exploring the roles of wild species as reservoirs of infectious diseases have become more common thanks to the technological breakthrough of high-throughput sequencing. Considering that some species are reservoirs of numerous viruses, including zoonotic ones, studies on viral diversity in rodents have recently increased [47][48][49][50][51][47-51]. However, few studies explored the links among viral diversity, host ecology, and habitats [28][52][53][28,52,53].
Here, we presented the viral diversity identified in three different organs of seven rodent species from French Guiana, Northern Amazonia, according to their natural hosts and habitats. The viromes were quantitatively dominated by vertebrate viral and to a lesser extent to viral sequences from invertebrates, plants, and amoeba. The different viral families, whether originating from invertebrates, plants, or vertebrates, were not evenly distributed within the different species and habitats. Viruses from Parvoviridae, Circoviridae, Astroviridae, and Anelloviridae from vertebrates were found in most species and habitats and can be considered as generalists. These ubiquitous viruses were already reported in wild rodents in United States [51], and Germany [54]. On the other hand, Caulimoviridae (from plants), Iflaviridae (from invertebrates), or Arteriviridae (from vertebrates) were rare and only present in some species and/or habitats. These differences in the distribution of viral families can be put in perspective by hypothesizing a rare biosphere for microbial diversity, with a portion of a few dominant microbial species and a second large, unexplored fraction with rare species [55]. Similarly, viromes in rodents could be dominated by a few dominant families, and a long distribution tail shaping a rare virosphere.
Such differences in virus abundance could be explained by the ecology of the viruses (ability to infect host cells, to persist and replicate) and by the ecology and behavior of rodent hosts in a given habitat. The role of vectors in viral transmission and their diversity according to the environment can also impact viral diversity. For example, for P. cuvieri and H. megacephalus, fourfold more viral families of invertebrate and vertebrate viruses have been detected in disturbed forest compared with pristine forest. In these two opportunistic species, diet can be supplemented by invertebrates when fruits and seeds are lacking [56], with subsequent impacts on their virome structures. On the other hand, a more specialized diet may restrict the range of viral diversity. Viral diversity indices and the relative dominance levels of viral species were also impacted by the level of disturbance and the type of habitat. The highest viral diversity index values were mainly observed in pristine habitats, where the highest diversity of hosts occur.
The virome of P. guyannensis in pristine forest showed the highest diversities compared to disturbed forest. This trend was nevertheless mitigated for P. cuvieri, although a higher diversity (α ≥ 0.25) and number of rare viral entities were observed in pristine forest.
In contrast, H. megacephalus presented a higher number of rare viruses in disturbed forests (high values of richness and at α = 0). Z. brevicauda, the only species sampled in savannas, showed the highest viral diversities in this habitat, also reflecting the richness of the savannah ecosystem [57][58][57,58]. The lowest viral diversity in peri-urban areas may be related to overall low biodiversity.
2.3.2. Lessons for Aassessing Eemergence Rrisks
The likelihood of disease emergence is commonly accepted to increase in disturbed habitats [59]. The transmission of viruses from forest species to humans may result from two mechanisms. First, anthropic activities can increase contact between wildlife and humans when enter in slightly modified habitats and come into contact with a pristine viral cycle, increasing the risk [25]. Secondly, in more degraded forests, environmental changes may disrupt some ecological barriers and impact the structure and dynamics of rodent and arthropod communities, species richness, and ecological networks [60]. This may favor generalist over specialist species and ultimately the dominance of more synanthropic ones. Feeding networks between hosts and hematophagous vectors consequently change, influencing the transmission of viruses and potentially increasing cross-species transmission events.
From a theoretical point of view, the dilution effect hypothesis explores how the decrease of biodiversity may increase the amplification of zoonotic diseases. Briefly, the dilution effect proposes that a high diversity of putative hosts and vector species dilutes the more efficient carriers and amplifiers of viruses in a community of less efficient species, consequently reducing the circulation of the harmful ones and lowering the likelihood of infection [61]. The dilution effect may affect cycles involving a single animal host (i.e., reservoir) and those with two host compartments, i.e., reservoirs and vectors. In the latter case, a decrease in vertebrate diversity may concentrate blood meals taken by arthropods on a lower number of species, resulting in a higher viral circulation as soon as those resilient vertebrate species are also efficient carriers. The dilution effect can be suggested to illustrate the links between the diversity of rodent hosts and the spread of some zoonotic viruses. A higher probability of hepacivirus infection in P. semispinosus has been related to a loss of diversity in hosts due to land-use change [62]. hantavirus outbreaks in the Americas are related to environmental disturbances that result in a decrease in specific richness of non-murine rodents and in the dominance of a few Muridae species known to be more efficient reservoirs [63][64][63,64]. In French Guiana, all known human hantavirus cases occurred in agricultural and peri-urban areas, where rodent diversity is much lower than in forest habitats [22], likely favoring hantavirus circulation in most efficient reservoirs.
2.3.3. Concluding Rremarks
Few studies on the viral diversity in rodents have been conducted, even though they comprise the first order of mammals in terms of the number of species and are considered an important source of viral zoonotic pathogens. In French Guiana, north of the Amazonian region, considered a hotspot of diversity for hosts and pathogens [65], the description of the virome of seven rodent species allowed identifying a large number of new viruses, most of which correspond to vertebrate viruses. These findings extend knowledge on the host range and evolution of these viruses. We showed that the diversity of rodent viromes varies according to the types of habitat, with higher viral diversity in pristine forests compared with disturbed forests for most rodent species. Environmental pressures on wild animal populations continue to grow, leading to increasing risks of contact between human and rodent populations. This could favor the emergence or re-emergence of viral diseases, including from viruses yet unknown or with undocumented roles on human health..
References
- Sanjuán, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral mutation rates. Virol. 2010, 84, 9733–9748, doi:10.1128/jvi.00694-10.
- Houldcroft, C.J.; Beale, M.A.; Breuer, J. Clinical and biological insights from viral genome sequencing. Rev. Microbiol. 2017, 15, 183–192, doi:10.1038/nrmicro.2016.182.
- Shi, M.; Lin, X.D.; Chen, X.; Tian, J.H.; Chen, L.J.; Li, K.; Wang, W.; Eden, J.S.; Shen, J.J.; Liu, L.; et al. The evolutionary history of vertebrate RNA viruses. Nature 2018, 556, 197–202, doi:10.1038/s41586-018-0012-7.
- Buck, C.B.; Van Doorslaer, K.; Peretti, A.; Geoghegan, E.M.; Tisza, M.J.; An, P.; Katz, J.P.; Pipas, J.M.; McBride, A.A.; Camus, A.C.; et al. The ancient evolutionary history of Polyomaviruses. PLoS Pathog. 2016, 12, e1000863, doi:10.1371/journal.ppat.1005574.
- Cobián Güemes, A.G.; Youle, M.; Cantú, V.A.; Felts, B.; Nulton, J.; Rohwer, F. Viruses as winners in the game of life. Rev. Virol. 2016, 3, 197–214, doi:10.1146/annurev-virology-100114-054952.
- Kreuder Johnson, C.; Hitchens, P.L.; Smiley Evans, T.; Goldstein, T.; Thomas, K.; Clements, A.; Joly, D.O.; Wolfe, N.D.; Daszak, P.; Karesh, W.B.; et al. Spillover and pandemic properties of zoonotic viruses with high host plasticity. Rep. 2015, 5, 1–8, doi:10.1038/srep14830
- Dennehy, J.J. Evolutionary ecology of virus emergence. N. Y. Acad. Sci. 2017, 1389, 124–146, doi:10.1111/nyas.13304.
- McMahon, B.J.; Morand, S.; Gray, J.S. Ecosystem change and zoonoses in the Anthropocene. Zoonoses Public Health 2018, 65, 755–765, doi:10.1111/zph.12489.
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global trends in emerging infectious diseases. Nature 2008, 451, 990–993, doi:10.1038/nature06536.
- Siu, A.; Wong, Y.C.R. Economic Impact of SARS: the case of Hong Kong. Asian Econ. Pap. 2004, 3, 62–83, doi:10.1162/1535351041747996.
- Huber, C.; Finelli, L.; Stevens, W. The economic and social burden of the 2014 Ebola outbreak in West Africa. Infect. Dis. 2018, 218, S698–S704, doi:10.1093/infdis/jiy213.
- European Centre for Disease Prevention and Control COVID-19 Situation Dashboard. Available online: https://qap.ecdc.europa.eu/public/extensions/COVID-19/COVID-19.html#global-overview-tabes (accessed on 20 July 2021).
- Bogich, T.L.; Chunara, R.; Scales, D.; Chan, E.; Pinheiro, L.C.; Chmura, A.A.; Carroll, D.; Daszak, P.; Brownstein, J.S. Preventing pandemics via international development: a systems approach. PLoS Med. 2012, 9, e1001354, doi:10.1371/journal.pmed.1001354.
- Available online: https://ohi.vetmed.ucdavis.edu/programs-projects/predict-project/about (accessed on 28 June 2021).
- Daszak, P.; Carroll, D.; Wolfe, N.; Mazet, J. The global virome project. J. Infect. Dis. 2016, 53, 36, doi:10.1016/j.ijid.2016.11.097.
- Carroll, D.; Daszak, P.; Wolfe, N.D.; Gao, G.F.; Morel, C.M.; Morzaria, S.; Pablos-Méndez, A.; Tomori, O.; Mazet, J.A.K. The Global Virome Project. Science (80) 2018, 359, 872–874, doi:10.1126/science.aap7463.
- Mollentze, N.; Streicker, D.G. Viral zoonotic risk is homogenous among taxonomic orders of mammalian and avian reservoir hosts. Natl. Acad. Sci. USA 2020, 117, 9423–9430, doi:10.1073/pnas.1919176117.
- Corbet, G.B.; Hill, J.E.; Wilson, D.E.; Reeder, D.M. (Eds.) Mammal Species Of the World: A Taxonomic and Geographic Reference, 2nd ed; Smithsonian Institution Press: Washington, DC, USA, 1993, ISBN 1-56098-217-9.
- Gravinatti, M.L.; Barbosa, C.M.; Soares, R.M.; Gregori, F. Synanthropic rodents as virus reservoirs and transmitters. Soc. Bras. Med. Trop. 2020, 53, 1–11, doi:10.1590/0037-8682-0486-2019.
- Han, B.A.; Schmidt, J.P.; Bowden, S.E.; Drake, J.M. Rodent reservoirs of future zoonotic diseases. Natl. Acad. Sci. USA 2015, 112, 7039–7044, doi:10.1073/pnas.1501598112
- Meerburg, B.G.; Singleton, G.R.; Kijlstra, A. Rodent-borne diseases and their risks for public health Rodent. Rev. Microbiol. 2009, 35, 221–270, doi:10.1080/10408410902989837.
- Lavergne, A.; Matheus, S.; Catzeflis, F.; Donato, D.; Lacoste, V.; de Thoisy, B. Rodent-borne viruses in French Guiana. Virologie 2017, 21, E12–E27, doi:10.1684/vir.2017.0697.
- Liu, J.; Liu, D.Y.; Chen, W.; Li, J.L.; Luo, F.; Li, Q.; Ling, J.X.; Liu, Y.Y.; Xiong, H.R.; Ding, X.H.; et al. Genetic analysis of hantaviruses and their rodent hosts in central-south China. Virus Res. 2012, 163, 439–447, doi:10.1016/j.virusres.2011.11.006.
- Monath, T.P.; Newhouse, V.F.; Kemp, G.E.; Setzer, H.W.; Cacciapuoti, A. Lassa virus isolation from Mastomys natalensis rodents during an epidemic in Sierra Leone. Science 1974, 185, 263–265, doi:10.1126/science.185.4147.263.
- Guégan, J.-F.F.; Ayouba, A.; Cappelle, J.; de Thoisy, B. Forests and emerging infectious diseases: unleashing the beast within. Res. Lett. 2020, 15, 083007, doi:10.1088/1748-9326/ab8dd7.
- Han, B.A.; Kramer, A.M.; Drake, J.M. Global patterns of zoonotic disease in mammals. Trends Parasitol. 2016, 32, 565–577, doi:10.1016/j.pt.2016.04.007.
- Allen, T.; Murray, K.A.; Zambrana-Torrelio, C.; Morse, S.S.; Rondinini, C.; Di Marco, M.; Breit, N.; Olival, K.J.; Daszak, P. Global hotspots and correlates of emerging zoonotic diseases. Commun. 2017, 8, 1–10, doi:10.1038/s41467-017-00923-8.
- Bergner, L.M.; Orton, R.J.; Benavides, J.A.; Becker, D.J.; Tello, C.; Biek, R.; Streicker, D.G. Demographic and environmental drivers of metagenomic viral diversity in vampire bats. Ecol. 2020, 29, 26–39, doi:10.1111/mec.15250
- Salmier, A.; Tirera, S.; de Thoisy, B.; Franc, A.; Darcissac, E.; Donato, D.; Bouchier, C.; Lacoste, V.; Lavergne, A. Virome analysis of two sympatric bat species (Desmodus rotundus and Molossus molossus) in French Guiana. PLoS ONE 2017, 12, e0186943, doi:10.1371/journal.pone.0186943.
- Bolatti, E.M.; Zorec, T.M.; Montani, E.; Hošnjak, L.; Chouhy, D.; Casal, P.E.; Barquez, M. American Free-Tailed Bats (Tadarida brasiliensis) and identification of two novel mammalian viruses. Viruses 2020, 12, 422.
- De Thoisy, B.; Matheus, S.; Catzeflis, F.; Clément, L.; Barrioz, S.; Guidez, A.; Donato, D.; Cornu, J.F.; Brunaux, O.; Guitet, S.; et al. Maripa Hantavirus in French Guiana: phylogenetic position and predicted spatial distribution of rodent hosts. J. Trop. Med. Hyg. 2014, 90, 988–992, doi:10.4269/ajtmh.13-0257.
- Matheus, S.; Kallel, H.; Mayence, C.; Bremand, L.; Houcke, S.; Rousset, D.; Lacoste, V.; de Thoisy, B.; Hommel, D.; Lavergne, A. Hantavirus pulmonary syndrome caused by Maripa virus in French Guiana, 2008–2016. Infect. Dis. 2017, 23, 1722–1725, doi:10.3201/eid2310.170842.
- Lavergne, A.; de Thoisy, B.; Donato, D.; Guidez, A.; Matheus, S.; Catzeflis, F.; Lacoste, V. Patawa Virus, a new Arenavirus hosted by forest rodents in French Guiana. Ecohealth 2015, 12, 339–346, doi:10.1007/s10393-014-0971-6.
- Lavergne, A.; de Thoisy, B.; Tirera, S.; Donato, D.; Bouchier, C.; Catzeflis, F.; Lacoste, V. Identification of lymphocytic choriomeningitis mammarenavirus in house mouse (Mus musculus, Rodentia) in French Guiana. Genet. Evol. 2016, 37, 225–230, doi:10.1016/j.meegid.2015.11.023.
- Sikes, R.S. 2016 Guidelines of the American Society of Mammalogists for the use of wild mammals in research and education. Mammal. 2016, 97, 663–688, doi:10.1093/jmammal/gyw078
- Tirera, S., de Thoisy, B., Donato, D., Bouchier, C., Lacoste, V., Franc, A., Lavergne, A. The influence of habitat on viral diversity in neotropical rodent hosts. Viruses 2021, 13, 1690.
- Lo, C.-C.; Chain, P.S.G. Rapid evaluation and quality control of next generation sequencing data with FaQCs. BMC Bioinform. 2014, 15, 366, doi:10.1186/s12859-014-0366-2
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2014, 31, 1674–1676, doi:10.1093/bioinformatics/btv033
- Li, D.; Luo, R.; Liu, C.M.; Leung, C.M.; Ting, H.F.; Sadakane, K.; Yamashita, H.; Lam, T.W. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 2016, 102, 3–11.
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv Prepr. arXiv 2013, arXiv:1303
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079, doi:10.1093/bioinformatics/btp352.
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. Mol. Biol. 1990, 215, 403–410, doi:10.1016/S0022-2836(05)80360-2.
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 1–9, doi:10.1186/1471-2105-10-421.
- Nayfach, S.; Camargo, A.P.; Schulz, F.; Eloe-Fadrosh, E.; Roux, S.; Kyrpides, N.C. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Biotechnol. 2020, 39, 578–585, doi:10.1038/s41587-020-00774-7.
- Hill, M.O. Diversity and evenness: an unifying notation and its consequences. Ecology 1973, 54, 427–432, doi:10.2307/1934352.
- Rényi, A. On measures of entropy and information. In Proceedings of the Fourth Berkeley Symposium on Mathematical Statistics and Probability; Contributions to the Theory of Statistics; University of California Press: Berkeley, CA, USA, 1961; Volume 1, pp. 547–561.
- Williams, S.H.; Che, X.; Garcia, J.A.; Klena, J.D.; Lee, B.; Muller, D.; Ulrich, W.; Corrigan, R.M.; Nichol, S.; Jain, K.; et al. Viral diversity of house mice in New York City. MBio 2018, 9, e02101-19, doi:10.1128/mBio.01354-17.
- Wu, Z.; Lu, L.; Du, J.; Yang, L.; Ren, X.; Liu, B.; Jiang, J.; Yang, J.; Dong, J.; Sun, L.; et al. Comparative analysis of rodent and small mammal viromes to better understand the wildlife origin of emerging infectious diseases. Microbiome 2018, 6, 1–14.
- Wu, Z.; Han, Y.; Liu, B.; Li, H.; Zhu, G.; Latinne, A.; Dong, J.; Sun, L.; Du, J.; Zhou, S.; et al. Decoding the RNA viromes of rodent lungs provides new visions into the origin and evolution pattern of rodent-borne diseases in Mainland Southeast Asia. Microbiome 2020, 9, 1–19, doi:10.21203/rs.3.rs-17323/v1.
- Firth, C.; Bhat, M.; Firth, M.A.; Williams, S.H.; Frye, M.J.; Simmonds, P.; Conte, J.M.; Ng, J.; Garcia, J.; Bhuva, N.P.; et al. Detection of zoonotic pathogens and characterization of novel viruses carried by commensal Rattus norvegicus in New York city. MBio 2014, 5, e01933-14, doi:10.1128/mBio.01933-14.
- Phan, T.G.; Kapusinszky, B.; Wang, C.; Rose, R.K.; Lipton, H.L.; Delwart, E.L. The fecal viral flora of wild rodents. PLoS Pathog. 2011, 7, e1002218, doi:10.1371/journal.ppat.1002218.
- Campbell, S.J.; Ashley, W.; Gil-Fernandez, M.; Newsome, T.M.; Di Giallonardo, F.; Ortiz-Baez, A.S.; Mahar, J.E.; Towerton, A.L.; Gillings, M.; Holmes, E.C.; et al. Red fox viromes in urban and rural landscapes. Virus Evol. 2020, 6, veaa065, doi:10.1093/ve/veaa065.
- Geoghegan, J.L.; Giallonardo, F. Di; Wille, M.; Ortiz-Baez, A.S.; Costa, V.A.; Ghaly, T.; Mifsud, J.C.O.; Turnbull, O.M.H.; Bellwood, D.R.; Williamson, J.E.; et al. Host evolutionary history and ecology shape virome composition in fishes. bioRxiv 2020, doi:10.1101/2020.05.06.081505.
- Sachsenröder, J.; Braun, A.; Machnowska, P.; Ng, T.F.F.; Deng, X.; Guenther, S.; Bernstein, S.; Ulrich, R.G.; Delwart, E.; Johne, R. Metagenomic identification of novel enteric viruses in urban wild rats and genome characterization of a group A rotavirus. Gen. Virol. 2014, 95, 2734–2747, doi:10.1099/vir.0.070029-0.
- Sogin, M.L.; Morrison, H.G.; Huber, J.A.; Welch, D.B.M.M.; Huse, S.M.; Neal, P.R.; Arrieta, J.M.; Herndl, G.J. Microbial diversity in the deep sea and the underexplored “rare biosphere. Natl. Acad. Sci. USA 2006, 103, 12115-12120, doi:10.1073/pnas.0605127103.
- Guillotin, M. Rythmes d'activité et régimes alimentaires de Proechimys cuvieri et d'Oryzomys capito velutinus (Rodentia) en forêt guyanaise. Ecol. (Terre Vie) 1982, 36, 337–371.
- Stier, A.; de Carvalho, W.D.; Rostain, S.; Catzeflis, F.; Claessens, O.; Dewynter, M.; McKey, D.; Mustin, K.; Palisse, M.; de Thoisy, B. The amazonian savannas of French Guiana: cultural and social importance, biodiversity, and conservation challenges. Conserv. Sci. 2020, 13, 1940082919900471, doi:10.1177/1940082919900471.
- Solbrig, O.T. The diversity of the savanna ecosystem. Stud. 1996, 121, 1–27, doi:10.1007/978-3-642-78969-4_1.
- Machalaba, C.; Karesh, W.B. Emerging infectious disease risk: shared drivers with environmental change. OIE Rev. Sci. Technol. 2017, 36, 435–444, doi:10.20506/rst.36.2.2664.
- Tilman, D.; May, R.M.; Lehman, C.L.; Nowack, M.A. Habitat destruction and the extinction debt revisited. Nature 1994, 371, 65–66, doi:10.2307/2269483.
- Ostfeld, R.S.; Keesing, F. Biodiversity and disease risk: the case of Lyme disease. Biol. 2000, 14, 722–728, doi:10.1046/j.1523-1739.2000.99014.x.
- Schmid, J.; Rasche, A.; Eibner, G.; Jeworowski, L.; Page, R.A.; Corman, V.M.; Drosten, C.; Sommer, S. Ecological drivers of Hepacivirus infection in a neotropical rodent inhabiting landscapes with various degrees of human environmental change. Oecologia 2018, 188, 289–302, doi:10.1007/s00442-018-4210-7.
- Ruedas, L.A.; Salazar-Bravo, J.; Tinnin, D.S.; Armién, B.; Cáceres, L.; García, A.; Díaz, M.A.; Gracia, F.; Suzán, G.; Peters, C.J.; et al. Community ecology of small mammal populations in Panamá following an outbreak of Hantavirus pulmonary syndrome. Vector Ecol. 2004, 29, 177–191.
- Suzán, G.; Marcé, E.; Giermakowski, J.T.; Mills, J.N.; Ceballos, G.; Ostfeld, R.S.; Armién, B.; Pascale, J.M.; Yates, T.L. Experimental evidence for reduced rodent diversity causing increased hantavirus prevalence. PLoS ONE 2009, 4, e5461, doi:10.1371/journal.pone.0005461.
- Lorenz, C.; de Oliveira Lage, M.; Chiaravalloti-Neto, F. Deforestation hotspots, climate crisis, and the perfect scenario for the next epidemic: The Amazon time bomb. Total Environ. 2021, 783, 147090, doi:10.1016/j.scitotenv.2021.147090