Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 4 by Vivi Li and Version 5 by Conner Chen.
The skin functions as a barrier between the organism and the surrounding environment. Direct exposure to external stimuli and the accumulation of genetic mutations may lead to abnormal cell growth, irreversible tissue damage and potentially favor skin malignancy. Skin homeostasis is coordinated by an intricate signaling network, and its dysregulation has been implicated in the development of skin cancers. Wnt signaling is one such regulatory pathway orchestrating skin development, homeostasis, and stem cell activation. Aberrant regulation of Wnt signaling cascades not only gives rise to tumor initiation, progression and invasion, but also maintains cancer stem cells which contribute to tumor recurrence.
- Wnt signaling
- non-melanoma skin cancer
- basal cell carcinoma
- squamous cell carcinoma
1. Introduction
The skin is the largest organ and functions as a protective barrier for the host to prevent fluid loss and regulate body temperature. It also harbors vasculature and sensory organs that transduce changes in temperature and pressure. The skin is composed of two major layers, the epidermis and dermis, forming the outer and inner layers, respectively. The epidermis includes the stratified epithelium, referred to as the interfollicular epidermis (IFE), and skin appendages, including the hair follicle, sebaceous gland, and sweat gland. The IFE and the hair follicle undergo regeneration throughout life. Constant exposure to mutagens, i.e., ultraviolet (UV) light and chemicals, can induce genetic mutations and hyperproliferation of epidermal cells, both of which eventually contribute to the formation of skin cancer. Two major types of non-melanoma skin cancer (NMSC) commonly found in patients are basal cell carcinoma (BCC) and squamous cell carcinoma (SCC), which together account for more than 95% of total NMSC [1]. As BCC and SCC share lineage with epidermal keratinocytes, they are specifically referred as keratinocyte carcinomas [2][3]. Although the risk of death from keratinocyte carcinomas is moderate, it increases considerably if patients are immunocompromised. Thus, identifying the drivers that induce skin tumors and understanding the molecular mechanisms responsible for tumor progression and maintenance is critical for biomarker discovery in diagnosis, prognosis, and therapy monitoring.
Several signaling pathways that have been shown to play vital roles in development of the skin epithelium are also implicated in the progression of keratinocyte carcinoma, including hedgehog (Hh), transforming growth factor β (TGFβ), mitogen-activated protein kinase (MAPK/ERK) and Wnt signaling [4][5][6][7]. Among them, Wnt signaling is of particularly interest due to its complexity in intracellular signaling cascades triggered by differential Wnt ligand-receptor combinations. Divergent roles of Wnt signaling have been discovered in multiple mammalian tissues during development and homeostasis of adult tissues. Canonical Wnt/β-catenin signaling is a major regulatory pathway that governs developmental processes as well as regulating maintenance and differentiation of adult stem cells (SCs) [8][9][10]. In this review, we summarize recent studies where the role of Wnt signaling in regulating tumor initiation and progression of keratinocyte carcinoma has been identified.
2. Wnt Signaling in Keratinocyte Carcinomas
While genetic mutations are major factors for the development of keratinocyte carcinoma, disrupted signaling pathways have emerged as necessary cofactors. Disruption of HF homeostasis may lead to hair loss disorders, such as alopecia universalis, or uncontrolled HF proliferation, which may cause follicle-based tumors [11]. Notably, aberrant activation of Wnt/β-catenin signaling is considered as one of the main driving elements causing developmental defects and tumorigenesis [12]. The constitutive expression of stabilized β-catenin in skin epidermis (K14-∆Nβ-catenin) causes the development of pilomatricomas or trichofolliculomas [13][14]. In contrast, depletion of β-catenin in carcinogen-induced SCCs results in tumor regression [15], indicating the essential role of β-catenin-dependent signaling in tumorigenesis. Genome-wide RNA-interference (RNAi) screening in the developing skin epidermis has reinforced the notion that β-catenin contributes to oncogenic growth [16]. Furthermore, in addition to the TCF/LEF transcription factor, nuclear β-catenin can also bind to the vitamin D receptor (VDR), which was shown to play a role in HF maintenance [17][18]. Inhibition of β-catenin-dependent transcriptional activity by overexpressing N-terminally truncated Lef1 in mouse epidermis (K14-∆NLef1) leads to the development of sebaceous gland (SG) tumors [19][20]. Interestingly, activation of β-catenin signaling (K14-∆Nβ-cateninER) in the absence of VDR causes development of tumors resembling BCCs [21]. These findings suggest that skin tumor types are specified by the interaction between β-catenin and its transcriptional effectors. Moreover, oncogenic activation of β-catenin in different epidermal SC populations results in distinct tumor types [22], implicating skin tumor heterogeneity. In addition to canonical Wnt signaling, non-canonical Wnt signaling is also implicated in epithelial-to-mesenchymal transition (EMT), a process involved in tumor metastasis and chemo-repulsion [23][24]. In the following sections, we focus on BCC and SCC, and discuss the implications of the Wnt signaling pathways in tumor formation and progression of these tumors.2.1. Basal Cell Carcinoma (BCC)
BCC is the most common skin tumor in humans. The main etiological factors provoking the development of BCCs are UV radiation, ionizing radiation, arsenic exposure, as well as traumatic injury or burn [25][26]. Other factors, such as wounding, can increase the risk of BCC development and malignancy in humans and mice [27][28]. It is noteworthy that BCCs are readily treated by means of various surgical methods at an early stage [29], and in some exceptional cases, BCC is reported to undergo self-regression [30][31][32]. However, once these lesions progress from BCC in situ to an advanced state, they are no longer amenable to surgery or radiation therapy. In even more rare cases, the tumor cells spread to distant sites (metastatic BCC). Although metastatic rate (<0.1%) and mortality caused by BCC is low, it may create substantial damage to skin tissue or morbidity if neglected for prolonged periods [33][34][35].
BCCs can be clinically and histologically categorized into several encompassing nodular, micronodular, superficial, infiltrative, morpheiform, and mixture variants. Thus, BCC is generally characterized based on the structure of tumor cells similar to the basal cells of the normal epidermis, however, their molecular characteristics are more related to embryonic hair follicle progenitors [36]. Recently, a new molecular classification was introduced apart from clinical and histopathological classifications. Based on the genomic profiling, BCCs are divided into three subtypes: 1) classical BCCs, which are closely associated with the Wnt and Hh signaling pathways; 2) normal-like BCCs, notably displaying an active fatty acid metabolism; and lastly 3) SCC-like BCCs, relying on immune-response and oxidative stress-related genes [37]. Here, we mainly focus on classical BCCs.
There is extensive evidence that the origin of BCC pathogenesis is predominantly triggered by dysregulation of the Hh pathway [38][39]. This could be attributable to aberrant genetic alterations that inactivate Patched 1 (PTCH1) or Suppressor of Fused (SUFU), constitutively activate Smoothened (SMO), or lead to overexpression of Glioma associated oncogene homolog 1 (GLI1) [4][26][40]. In the absence of Hh, Ptch acts to prevent activity of Smo. When Hh binds to and inhibits Ptch, Smo is activated to release inhibition of Gli by SuFu and kinesin family member 7 (Kif7), thus allowing Gli to enter the nucleus and initiate transcriptional activation of genes that regulate cell survival, cell cycle regulation and angiogenesis (Figure 1) [41][42][43]. Uncontrollable activation of Hh signaling prominently promotes tumorigenesis in sporadic and inherited BCCs. Based on recent genomic analysis, loss of PTCH1 and gain of SMO were described as causative mutant Hh pathway genes, accounting for 90% of human BCCs [26][44]. Mouse models of BCC genesis mainly rely on the repression of Patch1 or overexpression of Gli1/2. For example, mice overexpressing Gli1 in skin epidermis (K5-Gli1) develop several types of skin tumors, primarily hair follicle-derived tumors and BCCs [45], whereas Gli2 overexpression (K5-Gli2) only causes the formation of BCCs [46]. Thus, alterations in the expression of Hh signaling components may lead to the development of different tumor types. Moreover, the original cell populations in skin epidermis expressing excess Hh signaling also determine the phenotype of developed BCCs. For instance, over-activation of Gli2 in IFE gives rise to superficial BCC-like tumors, whereas HFSCs overexpressing Gli2 develop nodular BCC-like tumors [47]. Apart from IFE and HFs, innervated progenitors within mechanosensory niches were shown to be another plausible cell population that contributes to the development of BCCs [48].
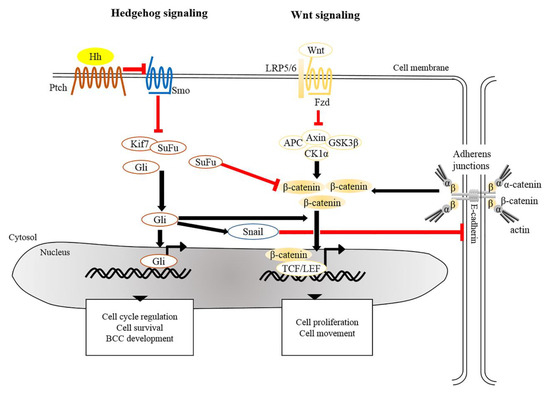
Figure 1. Crosstalk of signaling pathways in the pathogenesis of BCC. The main driver of BCC development is the dysregulation of Hedgehog signaling and Wnt/β-catenin signaling whereby the PTCH, SMO, SUFU, and CTNNB1 (a gene encoding for β-catenin) are frequently mutated in human BCCs. Mutated Ptch loses its grip on Smo that subsequently leads to its activation. Aberrant activation of Smo releases inhibition of Gli from SuFu and Kif7, thus allowing nuclear translocation of Gli. Aberrant Gli activity induces the expression of genes that regulate the cell cycle, cell survival and development of BCC. In addition, Gli induces Snail leading to the inhibition of E-cadherin, which in turn results in the accumulation of free β-catenin and its translocation to the nucleus. Alternatively, inactivation of SuFu and Kif7 leads to the accumulation and nuclear translocation of stabilized β-catenin which in turn facilitates BCC genesis. BCC, basal cell carcinoma; Gli, Glioma associated oncogenic homolog; Kif7, kinesin family member 7; Ptch, Patched; Smo, Smoothened; SuFu, Suppressor of Fused.
The development and progression of tumors are orchestrated by a network of intricate signaling pathways. In addition to Hh signaling, ample scientific evidence indicates that the Wnt/β-catenin signaling pathway participates in tumorigenesis of BCCs. Of note, constitutive expression of Wnt mediators, e.g., Wnt1, 2, 5A, 11, 13, and 16 and β-catenin, facilitates the progression of BCCs [49][50]. As reported previously, nearly 30% of human BCC samples exhibited an accumulation of β-catenin in the nucleus [51][52], and nuclear β-catenin in BCCs mainly resides at the tumor periphery [53]. In addition, BCCs which exhibit nuclear β-catenin display significantly higher proliferation rates [54]. Along this line, 25% of BCC samples contain the missense mutations in the third exon of the β-catenin gene [55], which is considered as a notable characteristic of hair follicle-related skin carcinoma [14][56]. Mutations in exon 3 of CTNNB1 (the gene encoding for β-catenin), particularly at Ser 33, 37 and Thr 41, perturb the phosphorylation sites for GSK3β, which leads to stabilization of β-catenin and in turn elevates Wnt/β-catenin signaling in favor the event of tumorigenesis [56][57]. As mentioned earlier, activation of β-catenin in the absence of VDR in mouse epidermis results in the development of undifferentiated tumors resembling BCCs [21]. Moreover, transcriptional profiling of adult epidermis expressing constitutively active SmoM2 shows that adult tumor-initiating cells are reprogramed into an embryonic hair follicle progenitor-like fate, in which Wnt/β-catenin signaling is highly activated. Depletion of β-catenin in adult epidermis expressing SmoM2 prevents embryonic reprograming and skin tumorigenesis [36]. Indeed, during skin development, β-catenin-dependent signaling directs the embryonic ectoderm to a HF-like fate [58][59], and over-activated β-catenin induces de novo HFs in adult epidermis [13]. Hence, β-catenin represents as a cardinal target in BCC arising from a HF-related origin.
Crosstalk between Hh and Wnt signaling pathways have been recently implicated in the pathogenesis of BCCs. Several genomic studies have indicated that genes encoding for components involved in both Hh and Wnt signaling are commonly altered in human BCCs [53][60]. It was shown that SuFu negatively regulates β-catenin signaling [61] and acts as a common regulator of Hh and Wnt signaling during Xenopus development [62]. Simultaneous inactivation of SuFu and Kif7 in adult epidermis results in the formation of BCCs that display increased nuclear β-catenin [63], reinforcing negative regulation of SuFu in β-catenin-dependent signaling. Moreover, overexpression of human GLI1 in frog epidermis induces BCC-like epidermal tumors which show specific upregulation of Wnt genes [50]. During epithelial transformation, Gli1 is able to induce the translocation of cytoplasmic β-catenin to the nucleus through modulation of E-cadherin [64]. Most recently, transcriptional profile of residual BCCs, which survive after treatment with Hh signaling inhibitor, reveals that Wnt signaling modulates cell identity of residual BCCs which may contribute to tumor relapse [65]. As a summary, in addition to the Hh pathway, dysregulation of Wnt/β-catenin signaling seems to be common during BCC development.
2.2. Squamous Cell Carcinoma (SCC)
SCC is recognized based on the tumor lesion composed of both proliferative basal cell and differentiated squamous cell layers. Epithelia arising from different parts of the body may develop different types of SCCs, including cutaneous SCC (cSCC), lung SCC (lSCC), and head and neck SCC (HNSCC). Each type of SCC has characteristics that can be distinguished by multidimensional genome-wide analyses [66]. One of the unique features for cSCC and HNSCC is their high degree of cellular heterogeneity [67][68][69]. As lSCC and HNSCC have been extensively discussed elsewhere [70][71][72], we mainly focus on cSCC in this review.
The incidence of cSCC is increasing remarkably each year due to a string of causative risk factors, including UV light from sun exposure, human papilloma virus (HPV) infection, chronic injury, arsenic exposure, immunosuppression, and inflammation [67][73]. Among these, UV light exposure is considered the most important accounting for 80–90% of identified cSCCs [74]. These prevailing factors promote the transformation of precancerous lesions, AK, to SCC in situ, invasive cSCC, and eventually metastatic SCC, as a consequence of multistep carcinogenesis. The malignancy of cSCC is favored by the accumulation of genetic and epigenetic alterations, viral pathogenesis, non-coding RNAs and dysregulation of signaling pathways [75]. The prognostic biomarkers of cSCCs can be determined by genetic, epigenetic, transcriptomic or proteomic analysis. These analyses provide invaluable information that hold the key for the selection of therapeutic intervention and for establishing comprehensive molecular landscapes of cSCC as future diagnostic biomarkers. The high rate of mortality and morbidity remains as a major concern attributed to the late diagnosis, ineffective treatment, relapse, and metastasis.
The main mutations of human cSCC are found in TP53, CDKN2A, RAS, PTEN, EGFR, and genes encoding for NOTCH receptors [75][76][77]. Around 75% of human cSCCs contain loss-of-function mutations in NOTCH1 or NOTCH2 [78]. In addition to the genes mentioned above, mutations of the telomerase reverse transcriptase (TERT) promoter are also found commonly in both human SCCs and BCCs [79][80]. Decades ago, in order to mimic SCC development in mice, a two-stage chemical carcinogenesis protocol, involving a single application of 7,12-Dimethylbenzanthracene (DMBA) followed by a repeated treatment with 12-O-Tetradecanoylphorbol-13-acetate (TPA), was exploited in murine models. DMBA administration causes the formation of DNA adducts, and successive treatment with TPA leads to sustained hyperplasia [81][82]. It is known that treatment with DMBA/TPA creates an accumulation of mutations in several critical genes, notably in H-Ras (A182T) [83], and provokes the progression of papilloma to malignant tumor, e.g., cSCC. Genetic alterations in genes encoding for RAS family members have been identified in a significant proportion of human tumors. More recently a genetic mouse model carrying oncogenic K-Ras(G12D) was widely utilized to study the process of tumorigenesis [84][85]. In adult epidermis, K-Ras(G12D) activation under the control of cell type-specific promoters allowed the origin for SCC formation to be defined. For instance, in combination with Tp53 deletion, K-Ras(G12D) activation in IFE basal cells or HFSCs, but not in transient amplifying HF matrix cells, led to the development of SCCs [85]. Although transformation by oncogenic RAS is an important event, RAS mutations only account for approximately 8–20% of human SCCs [76][86]. Additional mutations in tumor suppressor genes, e.g., TP53 or TP63 [6][87][88][89], are required to drive the malignancy of SCCs.
As previously described for BCCs, the cellular origin of SCCs affects the phenotype of the tumor. For instance, SCCs derived from the IFE are well-differentiated, whereas SCCs developing from the HFSCs frequently undergo EMT [90]. In addition to cell origin, the status of the originating cell also impacts on tumor formation. It has been shown that quiescent HFSCs are refractory to initiating tumors driven by activation of K-RAS(G12D) and Tp53 depletion [91]. Other than gene mutations, dysregulation of signaling pathways has also been implicated in mouse skin tumorigenesis [92]. In mouse SCCs, dysregulation of the PI3K/AKT signaling pathway, known to regulate a wide-range cellular functions, such as cell proliferation and apoptosis [93][94], is commonly found in RAS-induced tumors derived from HFSCs [6].
Dysregulation of Wnt signaling, including Wnt/β-catenin and β-catenin-independent Wnt/Ca2+ signaling, emerges as a major cause of cSCC development and progression [15][76][95]. Genetic alterations in Wnt-related ligands, receptors or mediators were identified in human cSCCs using various genomic profiling approaches. The first comparative genomic hybridization analysis indicated amplification of chromosome arms 7q, 8q, 11q, and 17q, which containing WNT and FZD genes, in cSCCs [96]. This finding was reinforced by microarray analysis revealing that WNT5A and FZD6 are both upregulated as unique gene signatures in human cSCCs [95][97]. Recently, genomic profiling of 122 human cSCC samples identified clinically relevant genomic alterations (CRGAs) showing that the key mutations in cSCCs not only include truncations of TP53 (85.2%), CDKN2A (61.5%), and NOTCH1 (42.6%), but also alterations in Wnt-related genes, LRP1B (22.1%) and APC (8.2%) [76]. In addition, another genomic study of cSCC supports the previous genomic profiling studies in which Wnt signaling was one of the common mutated pathways in human cSCCs [77].
The potential role of Wnt antagonists, SFRPs and Dkks, in tumorigenesis has drawn interest. Several lines of studies indicated that SFRP1, which destabilizes β-catenin by interfering with Wnt-Fzd interactions, was downregulated in human SCC samples [95]. Accordingly, hypermethylation of several SFRP genes, including SFRP1, 2, 4, and 5, have been identified in cSCC tumors and recognized as critical prognostic determinants for cSCCs [98]. Although hypermethylated SFRP genes could have caused hyper-activation of Wnt signaling that might contribute to cSCC, mechanisms of how SFRPs modulate skin cancer pathogenesis will require further investigation. In addition to SFRPs, Dkk proteins, known as negative regulators of canonical Wnt signaling, could also potentially serve as bona fide tumor suppressors. Accompanied by elevated β-catenin expression levels, downregulation of Dkk1 was seen in human cSCCs [99]. A decrease in Dkk3 expression was also detected in cSCCs. Overexpression of Dkk3 in SCC cells significantly reduced proliferation and migration [100], suggesting that Dkk3 has inhibitory effects on SCC development.
Aberrant accumulation of β-catenin protein or its presence in the nucleus in tumor cells is a common characteristic of SCCs [15][55][99][101][102]. Wnt/β-catenin signaling plays dual roles in both regulating normal SC self-renewal and maintenance and in cancer stem cells (CSCs). As discussed previously, depletion of β-catenin in carcinogen-induced SCCs halts tumor progression and eventually results in tumor regression, suggesting roles in maintenance of cutaneous CSCs [15]. This paradigm is in agreement with the idea that CSC-like properties in cSCC could be induced by aberrant expression of microRNA (miRNA) by activated canonical Wnt signaling [103]. Furthermore, it has been shown that tumor volume in a xenograft model of human cSCCs could be dampened upon β-catenin knockdown [16], underscoring the causal relationship between β-catenin and cell proliferation of cSCCs. These notions were based on a number of earlier studies showing that activation of β-catenin signaling, either by impairment of Notch [104], inactivation of Presenilin 1 [105] or activation of ROCK [106] caused an increase in the expression of cyclin D1 subsequently led to hyperproliferation (Figure 2). Conversely, aberrant expression of genes involved in cell cycle control can trigger the onset of skin tumorigenesis through activation of Wnt/β-catenin signaling. The cyclin-dependent kinase inhibitor 2A and 2B (CDNK2AB) locus, genes encoding for tumor suppressors p16 (INK4A), p14 (ARF), and p15 (INK4B) that inhibit cell cycle progression, is frequently lost in wide-range of tumors. A recent study indicated that loss of CDKN2ab allows development of SCC in the presence of active Wnt7b in a 129P2 mouse background [107]. Cell division cycle 20 (CDC20), another crucial cell cycle regulatory molecule, is commonly increased in cSCCs. Downregulation of CDC20 inhibits Wnt/β-catenin signaling, thereby suppressing the proliferation of cSCC cells and promoting apoptosis [108]. Taken together, these studies highlight the importance of Wnt signaling in regulating tumor cell proliferation and maintaining CSC phenotypes, both of which promote the SCC progression and aggressiveness.
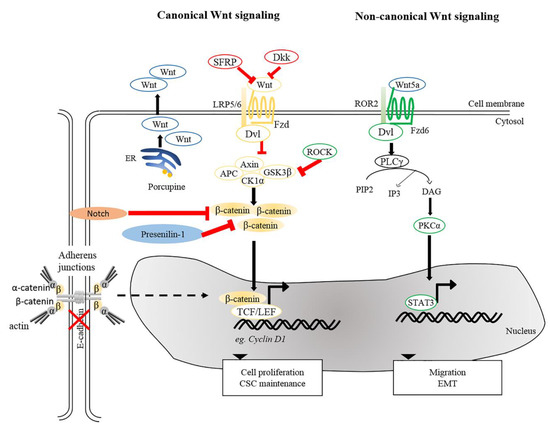
Figure 2. Wnt signaling pathways in cSCC. Canonical and non-canonical Wnt signaling participates in the maintenance of CSC, tumor progression, migration and EMT. Reduction of SFRPs and Dkks leads to activation of canonical Wnt signaling. Porcupine, an enzyme from the ER is needed for post-translational modification of Wnts to enable their transport and secretion. Other intricate factors, e.g., loss of E-cadherin or Presenillin-1, inhibition of Notch signaling and ROCK activation could modulate β-catenin signaling and activate genes involved in several cellular processes, including cell proliferation and CSC maintenance. For non-canonical Wnt signaling, interaction between Wnt5a and ROR2 facilitates EMT and invasive properties of cancer cells. Wnt5a is also required to activate PKCα and for STAT3 phosphorylation leading to tumorigenesis. ER, Endoplasmic reticulum; PKCα, protein kinase Cα; ROR2, receptor tyrosine kinase-like orphan receptor 2; STAT3, signal transducer and activator of transcription 3.
Apart from β-catenin, TCF7L1 (also known as TCF3), a binding partner for β-catenin, is known to contribute to tumor prevalence and progression in the pathogenesis of several cancers [109][110]. Motif-based analysis of human and mouse tissues identified TCF7L1 as one of the transcription factors whose targets are prominently altered during the progression from normal skin to SCCs [111]. A recent study provided evidence for a tumor-promoting role of TCF7L1 in skin. Overexpression of TCF7L1 increased tumor incidence, promoted tumor growth, and enhanced migration independent of β-catenin [112]. In addition to TCF7L1, Wnt genes are also commonly dysregulated in diverse tumor cells, including prostate, ovarian, colon, and skin cancers [113][114][115][116]. Upregulation of Wnt5a is clearly observed in human cSCC. Wnt5a is of major importance in maintaining the tumor phenotype in human SCCs [97][116][117], implying that Wnt5a serves as an oncogenic driver in skin cancer. It was reported that oncogenic signals mediated by Wnt5a were acquired via activation of PKCα and STAT3 phosphorylation, but not through Wnt/β-catenin pathway [117]. As mentioned above, immunosuppressed organ transplant recipients are more prone to SCCs [118]. One plausible factor is the elevated risk of HPV infection among immunosuppressed populations as compared to the immunocompetent patients [119]. Interestingly, vaccination against HPV successfully reduces the incidence of SCC in immunocompetent patients [120]. One study demonstrated that the presence of HPV induces the development of skin carcinomas in mice upon exposure to UV or wounding [121]. In addition, transcriptome analysis of HPV-driven mouse cSCCs indicated that transcripts encoding for Wnt ligands and Porcupine, a protein required for Wnt secretion, were highly upregulated in cSCCs. Inhibition of Porcupine by a small molecule inhibitor reduced the initiation and progression of HPV-driven cSCC [122].
In addition to cSCC maintenance and progression, β-catenin has an impact on tumor metastasis via EMT, in which E-cadherin-bound β-catenin is released from cell membranes and an increased pool of cytoplasmic β-catenin may enter the nucleus to trigger β-catenin-dependent signaling [123][124][125]. This notion is supported by studies showing that suppression of E-cadherin expression accelerates SCC progression by increasing invasion [126][127]. Besides canonical Wnt signaling, non-canonical Wnt ligand and receptor, Wnt5a, and ROR2, were shown to be required for Snail-mediated EMT and invasive properties of cancer cells [116]. In agreement, upregulated Wnt5 at the leading edge of SCCs suggests the contribution of Wnt5a-dependent signaling to tissue invasion by SCCs [128]. Together, these analyses and findings provide insights into the contribution of Wnt signaling to cSCC pathogenesis.
References
- Leiter, U.; Eigentler, T.; Garbe, C. Epidemiology of Skin Cancer. In Sunlight, Vitamin D and Skin Cancer; Springer: New York, NY, USA, 2014; pp. 120–140.
- Karimkhani, C.; Boyers, L.N.; Dellavalle, R.P.; Weinstock, M.A. It’s time for “keratinocyte carcinoma” to replace the term “nonmelanoma skin cancer”. J. Am. Acad. Dermatol. 2015, 72, 186–187.
- Nehal, K.S.; Bichakjian, C.K. Update on Keratinocyte Carcinomas. N. Engl. J. Med. 2018, 379, 363–374.
- Dahmane, N.; Lee, J.; Robins, P.; Heller, P.; Ruiz i Altaba, A. Activation of the transcription factor Gli1 and the Sonic hedgehog signalling pathway in skin tumours. Nature 1997, 389, 876–881.
- Cui, W.; Fowlis, D.J.; Bryson, S.; Duffie, E.; Ireland, H.; Balmain, A.; Akhurst, R.J. TGFbeta1 inhibits the formation of benign skin tumors, but enhances progression to invasive spindle carcinomas in transgenic mice. Cell 1996, 86, 531–542.
- White, A.C.; Tran, K.; Khuu, J.; Dang, C.; Cui, Y.; Binder, S.W.; Lowry, W.E. Defining the origins of Ras/p53-mediated squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2011, 108, 7425–7430.
- Watt, F.; Collins, C. Role of β-catenin in Epidermal Stem Cell Expansion, Lineage Selection, and Cancer. Cold Spring Harb. Symp. Quant. Biol. 2008, 73, 503–512.
- Holland, J.D.; Klaus, A.; Garratt, A.N.; Birchmeier, W.; Klaus-Bergmann, A. Wnt signaling in stem and cancer stem cells. Curr. Opin. Cell Biol. 2013, 25, 254–264.
- Van Amerongen, R.; Nusse, R. Towards an integrated view of Wnt signaling in development. Development 2009, 136, 3205–3214.
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999.
- Liu, S.; Zhang, H.; Duan, E. Epidermal Development in Mammals: Key Regulators, Signals from Beneath, and Stem Cells. Int. J. Mol. Sci. 2013, 14, 10869–10895.
- Klaus, A.; Birchmeier, W.; Klaus-Bergmann, A. Wnt signalling and its impact on development and cancer. Nat. Rev. Cancer 2008, 8, 387–398.
- Gat, U.; Dasgupta, R.; Degenstein, L.; Fuchs, E. De Novo hair follicle morphogenesis and hair tumors in mice expressing a truncated beta-catenin in skin. Cell 1998, 95, 605–614.
- Chan, E.; Gat, U.; McNiff, J.M.; Fuchs, E.; Chan, E.F. A common human skin tumour is caused by activating mutations in β-catenin. Nat. Genet. 1999, 21, 410–413.
- Malanchi, I.; Peinado, H.; Kassen, D.; Hussenet, T.; Metzger, D.; Chambon, P.; Huber, M.; Hohl, D.; Cano, A.; Birchmeier, W.; et al. Cutaneous cancer stem cell maintenance is dependent on beta-catenin signalling. Nature 2008, 452, 650–653.
- Beronja, S.; Janki, P.; Heller, E.; Lien, W.-H.; Keyes, B.E.; Oshimori, N.; Fuchs, E. RNAi screens in mice identify physiological regulators of oncogenic growth. Nature 2013, 501, 185–190.
- Shah, S.; Islam, M.N.; Dakshanamurthy, S.; Rizvi, I.; Rao, M.; Herrell, R.; Zinser, G.; Valrance, M.; Aranda, A.; Moras, D.; et al. The Molecular Basis of Vitamin D Receptor and β-Catenin Crossregulation. Mol. Cell 2006, 21, 799–809.
- Watt, F.M.; Celso, C.L.; Silva-Vargas, V. Epidermal stem cells: An update. Curr. Opin. Genet. Dev. 2006, 16, 518–524.
- Niemann, C.; Owens, D.M.; Hülsken, J.; Birchmeier, W.; Watt, F.M. Expression of DeltaNLef1 in mouse epidermis results in differentiation of hair follicles into squamous epidermal cysts and formation of skin tumours. Development 2002, 129, 95–109.
- Niemann, C.; Owens, D.M.; Schettina, P.; Watt, F.M. Dual Role of Inactivating Lef1 Mutations in Epidermis: Tumor Promotion and Specification of Tumor Type. Cancer Res. 2007, 67, 2916–2921.
- Palmer, H.G.; Anjos-Afonso, F.; Carmeliet, G.; Takeda, H.; Watt, F.M. The Vitamin D Receptor Is a Wnt Effector that Controls Hair Follicle Differentiation and Specifies Tumor Type in Adult Epidermis. PLoS ONE 2008, 3, e1483.
- Kretzschmar, K.; Weber, C.; Driskell, R.R.; Calonje, E.; Watt, F.M. Compartmentalized Epidermal Activation of β-Catenin Differentially Affects Lineage Reprogramming and Underlies Tumor Heterogeneity. Cell Rep. 2016, 14, 269–281.
- Gujral, T.S.; Chan, M.; Peshkin, L.; Sorger, P.K.; Kirschner, M.W.; MacBeath, G. A noncanonical Frizzled2 pathway regulates epithelial-mesenchymal transition and metastasis. Cell 2014, 159, 844–856.
- Galluzzi, L.; Spranger, S.; Fuchs, E.; López-Soto, A. WNT Signaling in Cancer Immunosurveillance. Trends Cell Biol. 2019, 29, 44–65.
- Rubin, A.I.; Chen, E.H.; Ratner, D. Basal-cell carcinoma. N. Engl. J. Med. 2005, 353, 2262–2269.
- Epstein, E.H. Basal cell carcinomas: Attack of the hedgehog. Nat. Rev. Cancer 2008, 8, 743–754.
- Kasper, M.; Jaks, V.; Are, A.; Bergström, Å.; Schwäger, A.; Svärd, J.; Teglund, S.; Barker, N.; Toftgård, R. Wounding enhances epidermal tumorigenesis by recruiting hair follicle keratinocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 4099–4104.
- Wong, S.Y.; Reiter, J.F. Wounding mobilizes hair follicle stem cells to form tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 4093–4098.
- Puig, S.; Berrocal, A. Management of high-risk and advanced basal cell carcinoma. Clin. Transl. Oncol. 2015, 17, 497–503.
- Curson, C.; Weedon, D. Spontaneous Regression in Basal Cell Carcinomas. J. Cutan. Pathol. 1979, 6, 432–437.
- Gupta, M.; Puri, P.; Kamal, A.; Nelson, M.E. Complete spontaneous regression of a basal cell carcinoma. Eye 2003, 17, 262–263.
- Fujimura, T.; Kakizaki, A.; Kambayashi, Y.; Aiba, S. Basal Cell Carcinoma with Spontaneous Regression: A Case Report and Immunohistochemical Study. Case Rep. Dermatol. 2012, 4, 125–132.
- Von Domarus, H.; Stevens, P.J. Metastatic basal cell carcinoma. Report of five cases and review of 170 cases in the literature. J. Am. Acad. Dermatol. 1984, 10, 1043–1060.
- Tilli, C.; Van Steensel, M.; Krekels, G.; Neumann, H.; Ramaekers, F. Molecular aetiology and pathogenesis of basal cell carcinoma. Br. J. Dermatol. 2005, 152, 1108–1124.
- Dourmishev, L.A.; Rusinova, D.; Botev, I. Clinical variants, stages, and management of basal cell carcinoma. Indian Dermatol. Online J. 2013, 4, 12–17.
- Youssef, K.K.; Lapouge, G.; Bouvrée, K.; Rorive, S.; Brohée, S.; Appelstein, O.; Larsimont, J.-C.; Sukumaran, V.; Van De Sande, B.; Pucci, D.; et al. Adult interfollicular tumour-initiating cells are reprogrammed into an embryonic hair follicle progenitor-like fate during basal cell carcinoma initiation. Nat. Cell Biol. 2012, 14, 1282–1294.
- Jee, B.A.; Lim, H.; Kwon, S.M.; Jo, Y.; Park, M.C.; Lee, I.J.; Woo, H.G. Molecular classification of basal cell carcinoma of skin by gene expression profiling. Mol. Carcinog. 2015, 54, 1605–1612.
- Rubin, L.L.; De Sauvage, F.J. Targeting the Hedgehog pathway in cancer. Nat. Rev. Drug Discov. 2006, 5, 1026–1033.
- Daya-Grosjean, L.; Couvé-Privat, S. Sonic hedgehog signaling in basal cell carcinomas. Cancer Lett. 2005, 225, 181–192.
- Reifenberger, J.; Wolter, M.; Knobbe, C.B.; Kohler, B.; Schönicke, A.; Scharwächter, C.; Kumar, K.; Blaschke, B.; Ruzicka, T.; Reifenberger, G. Somatic mutations in the PTCH, SMOH, SUFUH and TP53 genes in sporadic basal cell carcinomas. Br. J. Dermatol. 2005, 152, 43–51.
- Duman-Scheel, M.; Weng, L.; Xin, S.; Du, W. Hedgehog regulates cell growth and proliferation by inducing Cyclin D and Cyclin E. Nature 2002, 417, 299–304.
- Bigelow, R.L.; Chari, N.S.; Unden, A.B.; Spurgers, K.B.; Lee, S.; Roop, D.R.; Toftgard, R.; McDonnell, T.J. Transcriptional regulation of bcl-2 mediated by the sonic hedgehog signaling pathway through gli-1. J. Biol. Chem. 2004, 279, 1197–1205.
- Pola, R.; Ling, L.E.; Silver, M.; Corbley, M.J.; Kearney, M.; Pepinsky, R.B.; Shapiro, R.; Taylor, F.R.; Baker, D.P.; Asahara, T.; et al. The morphogen Sonic hedgehog is an indirect angiogenic agent upregulating two families of angiogenic growth factors. Nat. Med. 2001, 7, 706–711.
- Bonilla, X.; Parmentier, L.; King, B.; Bezrukov, F.; Kaya, G.; Zoete, V.; Seplyarskiy, V.B.; Sharpe, H.J.; McKee, T.; Letourneau, A.; et al. Genomic analysis identifies new drivers and progression pathways in skin basal cell carcinoma. Nat. Genet. 2016, 48, 398–406.
- Nilsson, M.; Undèn, A.B.; Krause, D.; Malmqwist, U.; Raza, K.; Zaphiropoulos, P.G.; Toftgård, R. Induction of basal cell carcinomas and trichoepitheliomas in mice overexpressing GLI-1. Proc. Natl. Acad. Sci. USA 2000, 97, 3438–3443.
- Grachtchouk, M.; Mo, R.; Yu, S.; Zhang, X.; Sasaki, H.; Hui, C.-C.; Dlugosz, A.A. Basal cell carcinomas in mice overexpressing Gli2 in skin. Nat. Genet. 2000, 24, 216–217.
- Grachtchouk, M.; Pero, J.; Yang, S.H.; Ermilov, A.N.; Michael, L.E.; Wang, A.; Wilbert, D.; Patel, R.M.; Ferris, J.; Diener, J.; et al. Basal cell carcinomas in mice arise from hair follicle stem cells and multiple epithelial progenitor populations. J. Clin. Investig. 2011, 121, 1768–1781.
- Peterson, S.C.; Eberl, M.; Vagnozzi, A.N.; Belkadi, A.; Veniaminova, N.A.; Verhaegen, M.E.; Bichakjian, C.K.; Ward, N.L.; Dlugosz, A.A.; Wong, S.Y. Basal cell carcinoma preferentially arises from stem cells within hair follicle and mechanosensory niches. Cell Stem Cell 2015, 16, 400–412.
- Muzio, L.L.; Pannone, G.; Staibano, S.; Mignogna, M.D.; Grieco, M.; Ramires, P.; Romito, A.M.; De Rosa, G.; Piattelli, A. WNT-1 expression in basal cell carcinoma of head and neck. An immunohistochemical and confocal study with regard to the intracellular distribution of beta-catenin. Anticancer Res. 2002, 22, 565–576.
- Mullor, J.L.; Dahmane, N.; Sun, T.; Ruiz i Altaba, A. Wnt signals are targets and mediators of Gli function. Curr. Biol. 2001, 11, 769–773.
- Yamazaki, F.; Aragane, Y.; Tezuka, T.; Kawada, A. Immunohistochemical detection for nuclear beta-catenin in sporadic basal cell carcinoma. Br. J. Dermatol. 2001, 145, 771–777.
- El-Bahrawy, M.; El-Masry, N.; Alison, M.; Poulsom, R.; Fallowfield, M. Expression of beta-catenin in basal cell carcinoma. Br. J. Dermatol. 2003, 148, 964–970.
- Brinkhuizen, T.; Hurk, K.V.D.; Winnepenninckx, V.J.L.; De Hoon, J.P.; Van Marion, A.M.; Veeck, J.; Van Engeland, M.; Van Steensel, M.A.M. Epigenetic Changes in Basal Cell Carcinoma Affect SHH and WNT Signaling Components. PLoS ONE 2012, 7, e51710.
- Saldanha, G.; Ghura, V.; Potter, L.; Fletcher, A. Nuclear beta-catenin in basal cell carcinoma correlates with increased proliferation. Br. J. Dermatol. 2004, 151, 157–164.
- Doglioni, C.; Piccinin, S.; Demontis, S.; Cangi, M.G.; Pecciarini, L.; Chiarelli, C.; Armellin, M.; Vukosavljevic, T.; Boiocchi, M.; Maestro, R. Alterations of beta-catenin pathway in non-melanoma skin tumors: Loss of alpha-ABC nuclear reactivity correlates with the presence of beta-catenin gene mutation. Am. J. Pathol. 2003, 163, 2277–2287.
- Kajino, Y.; Yamaguchi, A.; Hashimoto, N.; Matsuura, A.; Sato, N.; Kikuchi, K. Beta-Catenin gene mutation in human hair follicle-related tumors. Pathol. Int. 2001, 51, 543–548.
- Gao, C.; Wang, Y.; Broaddus, R.; Sun, L.; Xue, F.; Zhang, W. Exon 3 mutations of CTNNB1 drive tumorigenesis: A review. Oncotarget 2018, 9, 5492–5508.
- Zhang, Y.; Andl, T.; Yang, S.H.; Teta, M.; Liu, F.; Seykora, J.T.; Tobias, J.W.; Piccolo, S.; Schmidt-Ullrich, R.; Nagy, A.; et al. Activation of beta-catenin signaling programs embryonic epidermis to hair follicle fate. Development 2008, 135, 2161–2172.
- Järvinen, E.; Birchmeier, W.; Taketo, M.M.; Mikkola, M.L.; Thesleff, I.; Närhi, K. Sustained epithelial-catenin activity induces precocious hair development but disrupts hair follicle down-growth and hair shaft formation. Development 2008, 135, 1019–1028.
- Asplund, A.; Björklund, M.G.; Sundquist, C.; Strömberg, S.; Edlund, K.; Ostman, A.; Nilsson, P.; Pontén, F.; Lundeberg, J. Expression profiling of microdissected cell populations selected from basal cells in normal epidermis and basal cell carcinoma. Br. J. Dermatol. 2008, 158, 527–538.
- Meng, X.; Poon, R.; Zhang, X.; Cheah, A.; Ding, Q.; Hui, C.-C.; Alman, B. Suppressor of Fused Negatively Regulates β-Catenin Signaling. J. Boil. Chem. 2001, 276, 40113–40119.
- Min, T.H.; Kriebel, M.; Hou, S.; Pera, E.M. The dual regulator Sufu integrates Hedgehog and Wnt signals in the early Xenopus embryo. Dev. Boil. 2011, 358, 262–276.
- Li, Z.J.; Nieuwenhuis, E.; Nien, W.; Zhang, X.; Zhang, J.; Puviindran, V.; Wainwright, B.J.; Kim, P.C.W.; Hui, C.-C. Kif7 regulates Gli2 through Sufu-dependent and -independent functions during skin development and tumorigenesis. Development 2012, 139, 4152–4161.
- Li, X.; Deng, W.; Lobo-Ruppert, S.M.; Ruppert, J.M. Gli1 acts through Snail and E-cadherin to promote nuclear signaling by β-catenin. Oncogene 2007, 26, 4489–4498.
- Biehs, B.; Dijkgraaf, G.J.P.; Piskol, R.; Alicke, B.; Boumahdi, S.; Peale, F.; Gould, S.E.; De Sauvage, F.J. A cell identity switch allows residual BCC to survive Hedgehog pathway inhibition. Nature 2018, 562, 429.
- Gibb, E.A.; Enfield, K.S.; Tsui, I.F.; Chari, R.; Lam, S.; Alvarez, C.E.; Lam, W.L. Deciphering squamous cell carcinoma using multidimensional genomic approaches. J. Skin Cancer 2011, 2011, 541405.
- Alam, M.; Ratner, D. Cutaneous Squamous-Cell Carcinoma. N. Engl. J. Med. 2001, 344, 975–983.
- South, A.P.; Purdie, K.J.; Watt, S.A.; Haldenby, S.; Breems, N.D.; Dimon, M.; Arron, S.T.; Kluk, M.J.; Aster, J.C.; McHugh, A.; et al. NOTCH1 mutations occur early during cutaneous squamous cell carcinogenesis. J. Investig. Dermatol. 2014, 134, 2630–2638.
- Sánchez-Danés, A.; Blanpain, C. Deciphering the cells of origin of squamous cell carcinomas. Nat. Rev. Cancer 2018, 18, 549–561.
- Alamoud, K.; Kukuruzinska, M. Emerging Insights into Wnt/β-catenin Signaling in Head and Neck Cancer. J. Dent. Res. 2018, 97, 665–673.
- Noguti, J.; De Moura, C.F.G.; Hossaka, T.A.; Franco, M.; Oshima, C.T.F.; Dedivitis, R.A.; Ribeiro, D.A. The role of canonical WNT signaling pathway in oral carcinogenesis: A comprehensive review. Anticancer. Res. 2012, 32, 873–878.
- Rapp, J.; Jaromi, L.; Kvell, K.; Miskei, G.; Pongracz, J.E. WNT signaling—Lung cancer is no exception. Respir. Res. 2017, 18, 167.
- Dotto, G.P.; Rustgi, A.K. Squamous cell cancers: A unified perspective on biology and genetics. Cancer Cell 2016, 29, 622–637.
- Rigel, D.S. Cutaneous ultraviolet exposure and its relationship to the development of skin cancer. J. Am. Acad. Dermatol. 2008, 58, S129–S132.
- Tufaro, A.P.; Chuang, J.C.-M.; Prasad, N.; Chuang, A.; Chuang, T.C.; Fischer, A.C. Molecular Markers in Cutaneous Squamous Cell Carcinoma. Int. J. Surg. Oncol. 2011, 2011, 1–5.
- Al-Rohil, R.N.; Tarasen, A.J.; Carlson, J.A.; Wang, K.; Johnson, A.; Yelensky, R.; Lipson, D.; Elvin, J.A.; Vergilio, J.A.; Ali, S.M.; et al. Evaluation of 122 advanced-stage cutaneous squamous cell carcinomas by comprehensive genomic profiling opens the door for new routes to targeted therapies. Cancer 2016, 122, 249–257.
- Inman, G.J.; Wang, J.; Nagano, A.; Alexandrov, L.B.; Purdie, K.J.; Taylor, R.G.; Sherwood, V.; Thomson, J.; Hogan, S.; Spender, L.C.; et al. The genomic landscape of cutaneous SCC reveals drivers and a novel azathioprine associated mutational signature. Nat. Commun. 2018, 9, 3667.
- Wang, N.J.; Sanborn, Z.; Arnett, K.L.; Bayston, L.J.; Liao, W.; Proby, C.M.; Leigh, I.M.; Collisson, E.A.; Gordon, P.B.; Jakkula, L.; et al. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2011, 108, 17761–17766.
- Griewank, K.G.; Murali, R.; Schilling, B.; Schimming, T.; Möller, I.; Moll, I.; Schwamborn, M.; Sucker, A.; Zimmer, L.; Schadendorf, D.; et al. TERT Promoter Mutations Are Frequent in Cutaneous Basal Cell Carcinoma and Squamous Cell Carcinoma. PLoS ONE 2013, 8, e80354.
- Scott, G.A.; Laughlin, T.S.; Rothberg, P.G. Mutations of the TERT promoter are common in basal cell carcinoma and squamous cell carcinoma. Mod. Pathol. 2014, 27, 516–523.
- McCreery, M.Q.; Balmain, A. Chemical Carcinogenesis Models of Cancer: Back to the Future. Annu. Rev. Cancer Boil. 2017, 1, 295–312.
- Bigger, C.A.; Sawicki, J.T.; Blake, D.M.; Raymond, L.G.; Dipple, A. Products of binding of 7,12-dimethylbenz(a)anthracene to DNA in mouse skin. Cancer Res. 1983, 43, 5647–5651.
- Nelson, M.A.; Futscher, B.W.; Kinsella, T.; Wymer, J.; Bowden, G.T. Detection of mutant Ha-ras genes in chemically initiated mouse skin epidermis before the development of benign tumors. Proc. Natl. Acad. Sci. USA 1992, 89, 6398–6402.
- O’Hagan, R.C.; Heyer, J. KRAS Mouse Models: Modeling Cancer Harboring KRAS Mutations. Genes Cancer 2011, 2, 335–343.
- Lapouge, G.; Youssef, K.K.; Vokaer, B.; Achouri, Y.; Michaux, C.; Sotiropoulou, P.A.; Blanpain, C. Identifying the cellular origin of squamous skin tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 7431–7436.
- Boukamp, P. Non-melanoma skin cancer: What drives tumor development and progression? Carcinogenesis 2005, 26, 1657–1667.
- Brash, D.E.; Ziegler, A.; Jonason, A.S.; Simon, J.A.; Kunala, S.; Leffell, D.J. Sunlight and sunburn in human skin cancer: p53, apoptosis, and tumor promotion. J. Investig. Dermatol. Symp. Proc. 1996, 1, 136–142.
- Di Como, C.J.; Urist, M.J.; Babayan, I.; Drobnjak, M.; Hedvat, C.V.; Teruya-Feldstein, J.; Pohar, K.; Hoos, A.; Cordon-Cardo, C. p63 expression profiles in human normal and tumor tissues. Clin. Cancer Res. 2002, 8, 494–501.
- Ratushny, V.; Gober, M.D.; Hick, R.; Ridky, T.W.; Seykora, J.T. From keratinocyte to cancer: The pathogenesis and modeling of cutaneous squamous cell carcinoma. J. Clin. Investig. 2012, 122, 464–472.
- Latil, M.; Nassar, D.; Beck, B.; Boumahdi, S.; Wang, L.; Brisebarre, A.; Dubois, C.; Nkusi, E.; Lenglez, S.; Checinska, A.; et al. Cell-Type-Specific Chromatin States Differentially Prime Squamous Cell Carcinoma Tumor-Initiating Cells for Epithelial to Mesenchymal Transition. Cell Stem Cell 2017, 20, 191–204.e5.
- White, A.C.; Khuu, J.K.; Dang, C.Y.; Hu, J.; Tran, K.V.; Liu, A.; Gomez, S.; Zhang, Z.; Yi, R.; Scumpia, P.; et al. Stem cell quiescence acts as a tumour suppressor in squamous tumours. Nat. Cell Biol. 2014, 16, 99–107.
- Segrelles, C.; Ruiz, S.; Pérez, P.; Murga, C.; Santos, M.; Budunova, I.V.; Martínez, J.; Larcher, F.; Slaga, T.J.; Gutkind, J.S.; et al. Functional roles of Akt signaling in mouse skin tumorigenesis. Oncogene 2002, 21, 53–64.
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-Kinase–AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501.
- Claerhout, S.; Verschooten, L.; Van Kelst, S.; De Vos, R.; Proby, C.; Agostinis, P.; Garmyn, M. Concomitant inhibition of AKT and autophagy is required for efficient cisplatin-induced apoptosis of metastatic skin carcinoma. Int. J. Cancer 2010, 127, 2790–2803.
- Ra, S.H.; Li, X.; Binder, S. Molecular discrimination of cutaneous squamous cell carcinoma from actinic keratosis and normal skin. Mod. Pathol. 2011, 24, 963–973.
- Popp, S.; Waltering, S.; Herbst, C.; Moll, I.; Boukamp, P. UV-B-type mutations and chromosomal imbalances indicate common pathways for the development of Merkel and skin squamous cell carcinomas. Int. J. Cancer 2002, 99, 352–360.
- Haider, A.S.; Peters, S.B.; Kaporis, H.; Cardinale, I.; Fei, J.; Ott, J.; Blumenberg, M.; Bowcock, A.M.; Krueger, J.G.; Carucci, J.A.; et al. Genomic Analysis Defines a Cancer-Specific Gene Expression Signature for Human Squamous Cell Carcinoma and Distinguishes Malignant Hyperproliferation from Benign Hyperplasia. J. Investig. Dermatol. 2006, 126, 869–881.
- Liang, J.; Kang, X.; Halifu, Y.; Zeng, X.; Jin, T.; Zhang, M.; Luo, D.; Ding, Y.; Zhou, Y.; Yakeya, B.; et al. Secreted frizzled-related protein promotors are hypermethylated in cutaneous squamous carcinoma compared with normal epidermis. BMC Cancer 2015, 15, 58.
- He, X.; Li, S.; Luo, X.; Hu, D.; Cai, T.; Huang, K.; Zhou, W.; Chen, J. Expression of DKK1 and beta-catenin in epidermal neoplasms and their correlation. Int. J. Clin. Exp. Med. 2015, 8, 18843–18848.
- Shin, J.-M.; Choi, D.-K.; Kang, H.-Y.; Sohn, K.-C.; Lee, Y.; Kim, C.D.; Lee, J.-H.; Park, B.C. The expression pattern and functional role of REIC/Dkk-3 in the development of cutaneous squamous cell carcinoma. J. Dermatol. Sci. 2016, 84, 88–96.
- Lan, Y.J.; Chen, H.; Chen, J.Q.; Lei, Q.H.; Zheng, M.; Shao, Z.R. Immunolocalization of vimentin, keratin 17, Ki-67, involucrin, beta-catenin and E-cadherin in cutaneous squamous cell carcinoma. Pathol. Oncol. Res. 2014, 20, 263–266.
- Papadavid, E.; Pignatelli, M.; Zakynthinos, S.; Krausz, T.; Chu, A.C. Abnormal immunoreactivity of the E-cadherin/catenin (α-, β-, and γ-) complex in premalignant and malignant non-melanocytic skin tumours. J. Pathol. 2002, 196, 154–162.
- Bai, X.; Zhou, Y.; Chen, P.; Yang, M.; Xu, J. MicroRNA-142-5p induces cancer stem cell-like properties of cutaneous squamous cell carcinoma via inhibiting PTEN. J. Cell Biochem. 2018, 119, 2179–2188.
- Proweller, A.; Tu, L.; Lepore, J.J.; Cheng, L.; Lu, M.M.; Seykora, J.; Millar, S.E.; Pear, W.S.; Parmacek, M.S. Impaired Notch Signaling Promotes De novo Squamous Cell Carcinoma Formation. Cancer Res. 2006, 66, 7438–7444.
- Xia, X.; Qian, S.; Soriano, S.; Wu, Y.; Fletcher, A.M.; Wang, X.-J.; Koo, E.H.; Wu, X.; Zheng, H. Loss of presenilin 1 is associated with enhanced β-catenin signaling and skin tumorigenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 10863–10868.
- Samuel, M.S.; Lopez, J.I.; McGhee, E.J.; Croft, D.R.; Strachan, D.; Timpson, P.; Munro, J.; Schröder, E.; Zhou, J.; Brunton, V.G.; et al. Actomyosin-mediated cellular tension drives increased tissue stiffness and β-catenin activation to induce interfollicular epidermal hyperplasia and tumor growth. Cancer Cell 2011, 19, 776–791.
- Krimpenfort, P.; Snoek, M.; Lambooij, J.-P.; Song, J.-Y.; Van Der Weide, R.; Bhaskaran, R.; Teunissen, H.; Adams, D.J.; De Wit, E.; Berns, A. A natural WNT signaling variant potently synergizes with Cdkn2ab loss in skin carcinogenesis. Nat. Commun. 2019, 10, 1425.
- Chu, Z.; Zhang, X.; Li, Q.; Hu, G.; Lian, C.G.; Geng, S. CDC20 contributes to the development of human cutaneous squamous cell carcinoma through the Wnt/β-catenin signaling pathway. Int. J. Oncol. 2019, 54, 1534–1544.
- Murphy, M.; Chatterjee, S.S.; Jain, S.; Katari, M.; Dasgupta, R. TCF7L1 Modulates Colorectal Cancer Growth by Inhibiting Expression of the Tumor-Suppressor Gene EPHB3. Sci. Rep. 2016, 6, 28299.
- Zhang, B.; Wu, J.; Cai, Y.; Luo, M.; Wang, B.; Gu, Y. TCF7L1 indicates prognosis and promotes proliferation through activation of Keap1/NRF2 in gastric cancer. Acta Biochim. Biophys. Sin. 2019, 51, 375–385.
- Chitsazzadeh, V.; Coarfa, C.; Drummond, J.A.; Nguyen, T.; Joseph, A.; Chilukuri, S.; Charpiot, E.; Adelmann, C.H.; Ching, G.; Nguyen, T.N.; et al. Cross-species identification of genomic drivers of squamous cell carcinoma development across preneoplastic intermediates. Nat. Commun. 2016, 7, 12601.
- Ku, A.T.; Shaver, T.M.; Rao, A.S.; Howard, J.M.; Rodriguez, C.N.; Miao, Q.; Garcia, G.; Le, D.; Yang, D.; Borowiak, M.; et al. TCF7L1 promotes skin tumorigenesis independently of β-catenin through induction of LCN2. eLife 2017, 6, 6.
- Ricken, A.; Lochhead, P.; Kontogiannea, M.; Farookhi, R. Wnt Signaling in the Ovary: Identification and Compartmentalized Expression of wnt-2, wnt-2b, and Frizzled-4 mRNAs. Endocrinology 2002, 143, 2741–2749.
- Bakker, E.R.; Das, A.M.; Helvensteijn, W.; Franken, P.F.; Swagemakers, S.; Van Der Valk, M.A.; Hagen, T.L.T.; Kuipers, E.J.; Van Veelen, W.; Smits, R. Wnt5a promotes human colon cancer cell migration and invasion but does not augment intestinal tumorigenesis in Apc 1638N mice. Carcinogenesis 2013, 34, 2629–2638.
- Sandsmark, E.; Hansen, A.F.; Selnaes, K.M.; Bertilsson, H.; Bofin, A.M.; Wright, A.J.; Viset, T.; Richardsen, E.; Drablos, F.; Bathen, T.F.; et al. A novel non-canonical Wnt signature for prostate cancer aggressiveness. Oncotarget 2017, 8, 9572–9586.
- Ren, D.; Minami, Y.; Nishita, M. Critical role of Wnt5a-Ror2 signaling in motility and invasiveness of carcinoma cells following Snail-mediated epithelial-mesenchymal transition. Genes Cells 2011, 16, 304–315.
- Kang, M.-I.; Baker, A.R.; Dextras, C.R.; Cabarcas, S.M.; Young, M.R.; Colburn, N.H. Targeting of Noncanonical Wnt5a Signaling by AP-1 Blocker Dominant-Negative Jun When It Inhibits Skin Carcinogenesis. Genes Cancer 2012, 3, 37–50.
- Connolly, K.; Manders, P.; Earls, P.; Epstein, R.J. Papillomavirus-associated squamous skin cancers following transplant immunosuppression: One Notch closer to control. Cancer Treat. Rev. 2014, 40, 205–214.
- Harwood, C.A.; Surentheran, T.; McGregor, J.M.; Spink, P.J.; Leigh, I.M.; Breuer, J.; Proby, C.M. Human papillomavirus infection and non-melanoma skin cancer in immunosuppressed and immunocompetent individuals. J. Med Virol. 2000, 61, 289–297.
- Nichols, A.J.; Allen, A.H.; Shareef, S.; Badiavas, E.V.; Kirsner, R.S.; Ioannides, T. Association of Human Papillomavirus Vaccine with the Development of Keratinocyte Carcinomas. JAMA Dermatol. 2017, 153, 571–574.
- Marcuzzi, G.P.; Hufbauer, M.; Kasper, H.U.; Weissenborn, S.J.; Smola, S.; Pfister, H. Spontaneous tumour development in human papillomavirus type 8 E6 transgenic mice and rapid induction by UV-light exposure and wounding. J. Gen. Virol. 2009, 90, 2855–2864.
- Zimmerli, D.; Cecconi, V.; Valenta, T.; Hausmann, G.; Cantu, C.; Restivo, G.; Hafner, J.; Basler, K.; Broek, M.V.D. WNT ligands control initiation and progression of human papillomavirus-driven squamous cell carcinoma. Oncogene 2018, 37, 3753–3762.
- Grünert, S.; Jechlinger, M.; Beug, H.; Gruenert, S. Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat. Rev. Mol. Cell Boil. 2003, 4, 657–665.
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Boil. 2019, 20, 69–84.
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8.
- Margulis, A.; Zhang, W.; Alt-Holland, A.; Crawford, H.C.; Fusenig, N.E.; Garlick, J.A. E-cadherin Suppression Accelerates Squamous Cell Carcinoma Progression in Three-Dimensional, Human Tissue Constructs. Cancer Res. 2005, 65, 1783–1791.
- Heuberger, J.; Birchmeier, W. Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002915.
- Pourreyron, C.; Reilly, L.; Proby, C.; Panteleyev, A.; Fleming, C.; McLean, K.; South, A.P.; Foerster, J. Wnt5a Is Strongly Expressed at the Leading Edge in Non-Melanoma Skin Cancer, Forming Active Gradients, while Canonical Wnt Signalling Is Repressed. PLoS ONE 2012, 7, e31827.
More