Although COVID-19 pneumonia is a novel disease that is different from other types
of ARDS, severe COVID-19-associated ARDS shares typical ARDS lung pathology such
as diffuse alveolar damage and hyaline membrane formation. As Prasanna
et al. summarized, the general rationale for low-dose radiation treatment of COVID-19
is its inhibition of the cytokine storm, which promotes pulmonary dysfunction and ultimately ARDS. Inflammation is a dynamic and progressive process that is tightly associated with redox-modulated reactions. When recruited to sites of inflammation, macrophages and neutrophils generate reactive species, including reactive oxygen and nitrogen species (ROS and RNS). With multiple pro-inflammatory cytokines and chemokines being secreted, the latter together with elevated levels of ROS and RNS deteriorate redox homeostasis, and further worsen the disease. During the past two decades, research has revealed that low-dose radiation-mediated homeostasis is associated with enhanced cellular detoxification of ROS by a major antioxidant enzyme (manganese superoxide dismutase, MnSOD) within the mitochondria. This adaptive protection of mitochondrial metabolic functions is thought to provide experimental and theoretical support for using low-dose radiation to limit virus replication. Other antioxidants, including glutathione, were also shown to be increased following exposure to low doses of sparsely ionizing radiation such as X and γ rays. Schaue et al. suggested that it might be difficult and challenging for patients with complicated conditions and advanced age to rebalance redox levels, and low-dose radiation treatment might be of clinical value with its broad suppression of various inflammatory, pro-oxidant pathways at multiple levels.
- COVID-19
- SARS-CoV-2
- low-dose radiotherapy
- X-ray
1. Introduction
Humans have been living in a world affected by the novel coronavirus disease (COVID-19) for more than a year. Starting from a small group of infections, the highly infectious severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) viral strain has caused over 213 million reported infections, leading to ~4.46 million deaths so far [1]. Among the closed cases that had an outcome of either recovery and discharge or death, the case fatality rate is ~2% [1]. Although it has been argued that social measures have been largely ineffective [2], without the assistance of non-pharmaceutical interventions such as stay-at-home, use of sanitizers (hand washing), early case detection, and face masking, it is unlikely that the size of the outbreak can be mitigated or reduced [3]. Whereas active cases temporally declined in February and June 2021, they have rapidly risen again reaching an even higher record set in late April, and reaching over 744,000 daily new cases as of 19 August 2021 (among infected patients, 0.6% being in serious conditions) [1].
As an infectious disease, COVID-19 can be transmitted from human to human through respiratory droplets and aerosols created within a short distance, as well as prolonged interaction [4]. The World Health Organization (WHO) presented the “three Cs” as places with higher COVID-19 spreading risk: Crowded places with many people nearby, close-contact settings, where close-range conversations take place, and confined/enclosed spaces with poor ventilation [5]. These places include restaurants, bars, fitness facilities, theaters, and museums where people tend to be crowded and less fresh air is available. Thus, social distancing (at least 2 m apart and less than 30-min of close contact) has been advocated as a measure to reduce cluster transmission [6].
Similar to the seasonal flu, COVID-19 is a viral, respiratory illness. Whereas the flu results from an influenza virus infection, COVID-19 is caused by a new type of coronavirus— SARS-CoV-2 [7][8][7,8]. SARS-CoV-2 is one of the seven pathogenic coronaviruses that cause human respiratory diseases, which also include SARS-CoV, MERS-CoV (Middle-East respiratory syndrome coronavirus), HCoV-OC43 (human coronavirus OC43), HCoV-HKU1 (human coronavirus HKU1), HCoV-NL63 (human coronavirus NL63), and HCoV-229E (human coronavirus 229E) [9].
2. The Structure and Pathogenesis of SARS-CoV-2
1. Host cell attachment and receptor binding;
2. Host cell membrane and viral membrane fusion;
3. Viral genomic material residing in the host cell.
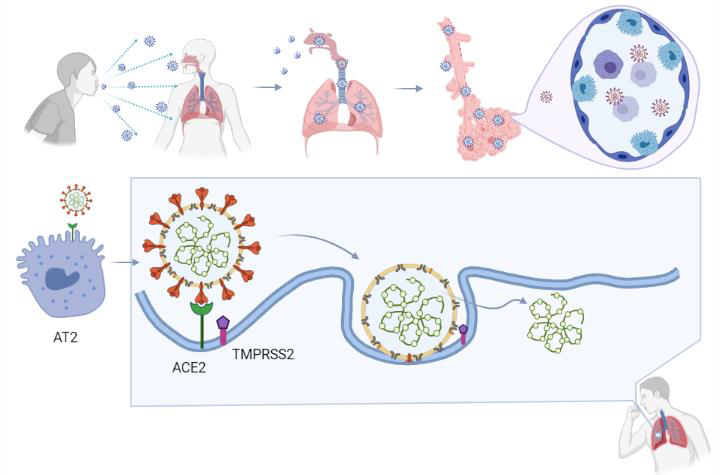
Coronaviruses employ a class 1 viral fusion protein, the homotrimeric S glycoprotein, which protrude from the viral surface to attach and bind to cell receptors [10][11][12][13][14][86,92,93,94,95]. SARS-CoV-2 S protein, in which a receptor-binding domain (RBD) structure is similar to that of SARS-CoV as it shares around 75% overall amino acid sequence identity, attracts ACE2 as a receptor to infect target cells [15][16][17][18][83,96,97,98]. ACE2 is not only highly expressed in lower respiratory tract cells such as type II alveolar cells (AT2) of the lung, but is found in organs such as the heart, kidney, and gastrointestinal tract, as well. This presumably contributes to COVID-19-induced ARDS, heart attack, kidney failure, and digestive syndromes [19][20][91,99]. During the infusion step, the S glycoprotein-ACE2 complex is proteolytically cleaved at a polybasic cleavage motif (PPAR) at the junction of two subunits of the spike protein (S1 and S2) by type 2 transmembrane serine protease (TMPRSS2) expressed on the surface of the host cell, leading to S protein activation and conformational alterations [21][22][23][24][100,101,102,103]. Similar to other coronaviruses, the S protein in SARS-CoV-2 is composed of two functional subunits, designated S1 and S2, of which the S1 subunit comprises the RBDs and stabilizes the prefusion state of the viral membrane-anchored S2 subunit [15][14][25][83,95,104]. Upon the dissociation of S1 from ACE2 by TMPRSS2, S2 undergoes a transition from a metastable pre-fusion state to a more stable post-fusion state, promoting the virus to enter target cells and release its genomic material into their cytoplasm [15][10][19][11][12][13][83,86,91,92,93,94]. Consistent with the sequencing map, S2 fusion machinery is more conserved than the S1 subunit, in line with the discovery that S1 is more exposed at the viral surface and is subject to more stringent pressure from the host immune system compared with the S2 subunit [15][25][83,104]. The sequence overall identity and structure conservation of non-RBD regions in the S protein such as the S2 subunit suggest potential targets for neutralizing antibodies and vaccines [15][10][83,86].
3. The immunological Changes Associated with COVID-19
When the viral infection starts (i.e., when SARS-CoV-2 reaches the cells that line the respiratory tract), the host responds by triggering the immune system to fight back. Although most patients recover successfully, a few but a non-negligible proportion of infected patients develop severe symptoms characterized by severe pneumonia, ARDS, lymphopenia, exhausted lymphocytes, cytokine storm, and possibly antibody-mediated enhancement of viral entry [26][27][28][47,119,120]. While the immune response attempts to defend against the infection, variable(s) yet to be clearly elucidated often counterbalance immune benefits leading patients to be hospitalized. Although the symptoms and outcomes are extraordinarily heterogeneous, understanding the relationship between disease severity and the various immunological changes that occur is under intense investigation [29][121].
In general, when humans are virally infected, specific cells produce interferons to block viruses from reproducing. Furthermore, the immune system marshals antibodies to target the invaders [30][122]. Specifically, IFN-γ (interferon-gamma)—a lymphokine secreted by activated T cells—directs and regulates the synthesis of IP-10 (IFN-γ-inducible protein-10), a protein involved in the inflammatory response [31][123]. IP-10 directs the recruitment of T-helper-1 (Th1) cells for both normal host defense against intracellular pathogens of the lung and acute and chronic inflammatory processes [32][124]. For example, studies in mice showed that a strong interferon response is crucial in the timely resolution of SARS-CoV and MERS-CoV infections. Alternatively, lethal inflammatory immune reactions are activated if the interferon response is delayed [33][34][125,126]. Significantly increased IP-10 levels have been shown in both SARS and MERS patients by independent groups over the past years [35][36][127,128].
In addition to the COVID-19 common symptoms such as fever and dry cough, bilateral patchy shadows, or ground-glass opacity on chest CT images, indicate lung damage. Together with CT scans that reveal damage to the lung, patients also present with high levels of IL-1B (interleukin-1B, an inflammatory cytokine), IFN-γ (T helper cytokine), IP-10, and MCP-1 (monocyte chemoattractant protein-1), which is similar to the association of these pro-inflammatory cytokines with pulmonary inflammation in SARS patients [26][37][47,129]. Depressed total lymphocytes counts (lymphopenia), prolonged prothrombin duration, and elevated lactate dehydrogenase were documented as the three most common abnormalities in hospitalized patients [38][46]. One of the early clinical reports in the city of Wuhan observed that compared to non-ICU patients, ICU patients had higher plasma levels of IL-2 (interleukin-2, a T helper cytokine), IL-7 (interleukin-7, a cytokine essential for lymphoid cell survival), IL-10 (interleukin-10, an anti-inflammatory cytokine), GCSF (granulocyte colony-stimulating factor), IP-10, MCP-1, MIP1α (macrophage inflammatory protein 1α, a small inducible cytokine involved in inflammatory responses), and TNF-α (tumor necrosis factor α, a pro-inflammatory cytokine) [26][39][40][47,130,131]. It was also found that white blood cell count, neutrophil count, the levels of D-dimer (indicator for blood vessel dysfunction and clot formation), blood urea, and creatinine levels continued to increase, while lymphocyte counts continued to decrease in non-surviving patients [38][41][46,132]. Despite COVID-19 being such a heterogeneous disease affecting patients with different ethnicities, ages, and underlying conditions, the triad of cytokines IP-10, IL-10, and IL-6 was identified to be severity-related, which might help in anticipating disease progression and the length of hospitalization [42][133].
4. Therapeutic Strategies
Hence, a cytokine storm, which is associated with high concentrations of pro-inflammatory cytokines in the plasma, is positively correlated with the severity of COVID-19 [26][47].
In a retrospective study of clinical treatment of hospitalized COVID-19 patients in early 2020, most patients received antiviral therapy (oseltamivir), many received antibacterial therapy (moxifloxacin, ceftriaxone, or azithromycin), and a few received glucocorticoid therapy when no specific treatment was recommended except supportive care in early 2020 [38][46]. Antibacterial drugs proved to be ineffective, and neither oseltamivir nor methylprednisolone (a strong corticosteroid) improved outcomes. Among those admitted into the ICU—patients who also often presented with hypertension, diabetes, cardiovascular disease, or cerebrovascular disease—high-flow oxygen therapy, non-invasive ventilation, and invasive ventilation (including extracorporeal membrane oxygenation as a rescue therapy) were applied according to specific individual situations [38][46].
During the early chaotic period of the COVID-19 pandemic, no antiviral drugs were efficacious until remdesivir showed limited improvement in shortening the recovery time in hospitalized adult patients [43][139]. Nonetheless, the cytokine storm-induced morbidity and mortality remained elevated [44][140]. Baricitinib, an inhibitor targeting the intracellular signaling pathway of cytokines, including IL-6, IL-10, IFN-γ, and GMCSF (granulocyte-macrophage colony-stimulating factor), improved patients’ quality of life. It also reduced conventional inflammatory parameters and IFN biomarkers (serum IP-10 level and 25-gene IFN score) in clinical trials treating monogenic IFN-mediated auto-inflammatory diseases [45][141]. Recently, the United States Food and Drug Administration (FDA), issued an emergency use of the anti-inflammatory drug baricitinib, in combination with the antiviral drug remdesivir, for the treatment of dysregulated inflammation for severe COVID-19 patients [46][142]. It was found that baricitinib plus remdesivir were superior to remdesivir alone in shortening the recovery time and improving clinical status with fewer serious adverse events in patients with COVID-19 [47][143].
Although the US FDA has authorized the emergency use of baricitinib in combination with remdesivir, additional investigation of treatment methods is needed due to the many confounding factors. The compensating effect of other key events in the involved molecular pathways is always a complicated issue for molecular targeting drugs. This is also the reason for clinical trials with various drug combinations, conducted in the hope of obtaining co-effective outcomes such as shortened recovery and reduced mortality. Considering the unbalanced immune conditions in severely ill patients, the mortality rate was not reduced significantly, although there was some improvement.