Allergic asthma is a chronic and heterogeneous pulmonary disease in which platelets can be activated in an IgE-mediated pathway and migrate to the airways via CCR3-dependent mechanism. Activated platelets secrete IL-33, Dkk-1, and 5-HT or overexpress CD40L on the cell surfaces to induce Type 2 immune response or interact with TSLP-stimulated myeloid DCs through the RANK-RANKL-dependent manner to tune the sensitization stage of allergic asthma. Additionally, platelets can mediate leukocyte infiltration into the lungs through P-selectin-mediated interaction with PSGL-1 and upregulate integrin expression in activated leukocytes.
- allergic asthma
- platelets
- adaptive immune response
- antiplatelet treatment
- megakaryocytes
1. Introduction
Asthma is a chronic and heterogeneous pulmonary disease which affects over 300 million people around the world [1]. Asthmatic patients can vary widely in terms of clinical presentation, severity, and pathophysiology, though they generally experience similar symptoms such as coughing, wheezing, breathlessness, and reversible airway obstruction. While its prevalence used to be reserved mainly for high-income countries, it has become a major public health challenge in China due to economic advances as well as rapid changes in environment and lifestyle [2]. Due to the complexity of the disease, there are several classification standards. Asthma phenotypes can be causatively grouped into allergic asthma and nonallergic asthma, or cellularly into eosinophilic, neutrophilic, paucigranulocytic, and mixed granulocytic asthma [3]. Furthermore, it is divided into Type 2 asthma and non-Type 2 asthma based on the inflammatory cytokine profiles [4,5][4][5].
Platelets are anucleate blood cells with a diameter of 2~4 μm generated from megakaryocytes. They contain a variety of secretory granules, such as α-granules, dense granules (δ-granules), and lysosomes, which not only contain coagulation-related factors, but also inflammatory mediators and protease [6]. Platelets are well known for their roles in hemostasis and thrombosis, and now increasing evidence has highlighted their immunological roles in inflammation ( Figure 1 ). Platelet malfunctions can lead to atherosclerosis, stroke, myocardial infarction, and deep venous thrombosis as well as allergic diseases, such as atopic dermatitis [7,8,9,10][7][8][9][10].
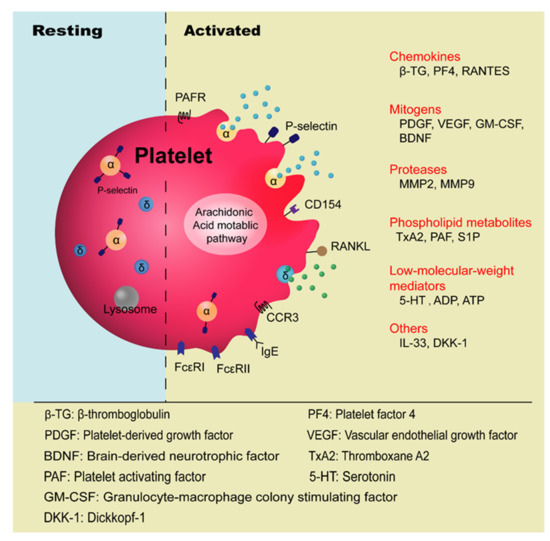
The involvement of platelets in the pathogenesis of asthma has been known for many years, especially in the development of allergic asthma and aspirin-exacerbated respiratory disease (AERD) [11,12][11][12]. Allergic asthma, also called atopic asthma, is the most common type of asthma with similar pathological features including mucus overproduction, chronic airway inflammation, airway remodeling, and airway hyperresponsiveness (AHR). It has been characterized as infiltration of eosinophils, mast cells, and lymphocytes in the airways and features increased secretion of type 2 cytokines such as IL-4, IL-5, and IL-13 after allergen exposure [13]. The onset of allergic asthma is influenced by both genetic backgrounds and environmental factors, and is closely related to allergic rhinitis [1,14][1][14]. Allergens that trigger allergic asthma include house dust mites, pollens, dander from cats and dogs, mold spores, cockroaches [15], and even small molecules such as toluene diisocyanate (TDI) [16].
2. Current Understanding of Platelets’ Involvement in Allergic Asthma
Platelet activation in allergic asthma has been highlighted by a great number of surveys ( Table 1 ). Disturbed hemostatic balance in the lungs of allergic asthma has been evidenced by increased levels of cellular fibronectin, a marker of vascular injury in asthmatic patients [17,18][17][18]. It is said that the imbalances between the coagulation and anticoagulation system and the fibrinolytic system are jointly involved in asthma [18]. The platelet activation markers such as β-thromboglobulin (β-TG) and platelet factor 4 (PF4), which are increased in the plasma of atopic dermatitis patients, were found to be higher when patients were afflicted with concomitant asthma and allergic rhinitis [10,19][10][19]. By intrabronchially challenging house-dust-mite (HDM)-sensitive asthmatic patients with Dermatophagoides pteronysisnus (Dp) extract, it has been shown that allergen challenge is correlated with platelet activation in vivo, manifested as decreased levels of platelet count and increased plasma levels of β-TG and PF4 [20]. The involvement of platelets in allergic asthma can be indicated by the increased platelet counts in the bronchoalveolar lavage fluid (BALF) of the patients and animal models [21]. Consistently, the increased levels of PF4 and β-TG were also detected in BALF [22]. Furthermore, platelet deposition on interalveolar septum walls was also observed in bronchial biopsies and its number was significantly increased after allergen challenge [23,24][23][24]. Moreover, platelet-leukocyte conjugates were also observed in the peripheral blood of asthmatic patients after allergen exposure [25]. Platelet activity in patients with pollen-induced seasonal allergic rhinitis and asthma, using plasma PF4 as an indicator, was found to increase during the grass pollen season and decrease in the off season [26]. It has been shown that P-selectin in the nasal lavage fluid of asthmatic patients is positively correlated with the level of eosinophil cationic protein (ECP) [27]. Furthermore, eosinophil β1-integrin activation in asthma was found to be associated with activated platelets in a P-selectin-mediated manner [28,29][28][29]. Duarte, D. et al. found a significantly higher level of platelet-derived microparticles (PMPs) in the peripheral blood of patients with allergic asthma compared to healthy individuals [30].
| Models | Samples | Indicators | Subjects | References |
|---|---|---|---|---|
| In vivo | Peripheral blood | Platelet-leukocyte conjugates↑ | Asthmatic patients after allergen exposure | [25] |
| Platelet-derived microparticles (PMPs) ↑ | Asthmatic patients | [30] | ||
| Percentage of IgE+ platelets↑ | Asthmatic patients | [31] | ||
| Eosinophil β1-integrin activation↑ | Asthmatic patients | [28][29] | ||
| Platelet BDNF↑ | Patients with allergic asthma | [32] | ||
| Plasma | β-TG and PF4↑ | Atopic dermatitis patients with concomitant asthma and allergic rhinitis | [19] | |
| β-TG, PF4↑ Platelet count↓ |
House-dust-mite-sensitive asthmatic patients intrabronchially challenged with Dp extract | [20] | ||
| PF4↑ (during the grass pollen season) PF4↓(off season) |
Patients with pollen-induced seasonal allergic rhinitis and asthma | [26] | ||
| BDNF↑ | Patients with allergic asthma | [32] | ||
| Phingosine-1-phosphate (SIP)↑ | House-dust-mite-allergic patients | [33] | ||
| BALF | Isolation of platelets | Asthmatic patients | [21] | |
| 5-HT↑ | Asthmatic patients | [31][34][35] | ||
| β-TG, PF4↑ | Ragweed-allergic asthmatic subjects after challenge with ragweed antigen. | [22] | ||
| Bronchial biopsies | Platelet deposition on interalveolar septum walls Platelet number↑ |
Asthmatic patients | [23][24][36] | |
| Nasal lavage fluid | P-selectin positively correlated with ECP level | Asthmatic patients | [27] | |
| In vitro | Platelet | FcεRI and FcεRII/CD23 expression | Human platelets and megakaryocyte | [31][34][35][37][38] |
| RANTES release↑ Cytotoxicity against schistosomula | Platelets treated by anti-FcεRI, anti-CD23, anti-IgE | [34][35][37] | ||
| RANTES release↑ | Platelet from allergic patients stimulated with IgE and anti-IgE | [35] | ||
| Allergen-specific cytotoxicity against schistosomula↑ | Patients allergic to Dermatophagoides pteronyssinus | [39] | ||
| Allergen-specific platelet chemotaxis | Allergic asthmatic patients | [38] | ||
| GM-CSF↑ Eosinophils apoptosis↓ |
Human platelets and Eosinophils coculture | [40] | ||
| P-selectin↑ Neutrophil superoxide anions↑ |
Human platelets and neutrophils coculture | [41] | ||
| RANKL in platelets↑ CCL17 (Th2-attracting chemokine)↑ |
TRAP6-activated platelets with TSLP-stimulated DCs coculture | [42] |
The expression of high-affinity IgE receptor (FcεRI) and low-affinity IgE receptors (FcεRII/CD23) on platelets provides the structural basis for platelets’ involvement in allergic asthma. Monoclonal antibodies targeting IgE receptors (anti-FcεRI and anti-CD23) or IgE binding to the asthmatic patients’ platelets (anti-IgE) induced the RANTES release by platelets and cytotoxicity against schistosomula [31,34,35,37][31][34][35][37]. It has also been demonstrated that the allergen triggered platelet activation in an IgE-FcεR-dependent pathway and induced inflammatory mediators, such as serotonin (5-HT) and RANTES, from platelets [34,35][34][35]. Research conducted on humans revealed that the percentage of IgE + platelets in atopic asthmatic patients was twice of that in humans with a normal IgE level, which is 10% [31]. This was also evidenced by the fact that the expression levels of FcεRI and FcεRII in platelets were significantly different between sham- and OVA-immunized mice [38]. However, others reported no distinct difference in the expression levels of FcεRI on the platelets in allergic patients and healthy controls [35].
In animal models, allergen exposure resulted in platelet migration to the airways, essential for eosinophil and lymphocyte recruitment and activation [25], and the critical role of P-selectin in platelets in the development of allergen-induced airway response was demonstrated in ovalbumin (OVA) and cockroach-induced murine asthmatic models [43,44][43][44]. Pitchford, S.C. et al. [38] showed that in OVA-sensitized mice, platelets migrated out of the blood vessels in an allergen-IgE-FcεRI pathway rather than due to hemorrhage after the allergen challenge. This migration of platelets occurred ahead of the infiltration of leukocytes into the lungs and was in single non-aggregated forms, which is different from the commonly observed aggregated state in thrombosis and hemostasis [38]. Due to the expression of chemokine receptors on the platelet surfaces, it is considered that they undergo chemotaxis in response to chemokines such as CCL11, CCL22, and CXCL12. Indeed, a recent study showed that CCR3 (CCL11 receptor) is essential for the recruitment of platelets into asthmatic lungs in the models of allergic inflammation. Under intravital microscopy, the rolling, adhesion, and extravascular migration of platelets in the HDM-sensitized murine models were markedly suppressed by SB328437, a CCR3 antagonist [36]. A study on platelet degranulation function in hemostasis and inflammation revealed that AHR and eosinophilic inflammation were significantly diminished in platelet-specific Munc13-4 KO mice, as the dense granule release was abrogated in these mice [45]. This evidence shows that platelets actively participate in the development of allergic asthma.
It is worth noting that while some have argued that allergens directly activate platelets based on the platelets’ anti-parasite cytotoxicity, others could not detect allergen-induced platelet aggregation and degranulation [39,46][39][46]. These contradicting results indicate that the underlying mechanism of platelets’ response to allergen is yet to be clarified. Interestingly, the work by Kasperska-Zajac, A. et al. found no significant differences in the plasma levels of PF-4 and β-TG in HDM-allergic patients and seasonal allergic rhinitis patients versus healthy non-atopic subjects [47,48][47][48]. Contrary to the notion that the vascular endothelial growth factor (VEGF) level is increased in plasma of allergic patients with asthma and atopic dermatitis, the work by Koczy-Baron, E. et al. showed that the free circulating VEGF level did not change in patients with chronic allergic rhinitis [49]. These discrepancies indicate that differences may exist in the platelet activity among patients with distinct clinical manifestations of atopy. This intriguing phenomenon was discussed in the review by Potaczek, D.P. [50].
3. Mechanisms of Platelets’ Role in the Pathogenesis of Allergic Asthma
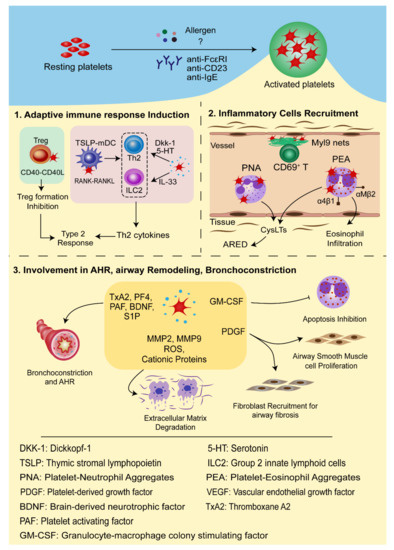
It is now widely accepted that the intrapulmonary migration of platelets is a critical process for the development of allergic asthma because it not only promotes the recruitment of eosinophils and other leukocytes from the blood vessels into lung tissues but also serves as a reservoir of inflammatory mediators to promote the pathogenic process of allergic asthma [38]. In this section, we will discuss how intrapulmonary platelets contribute to the pathogenesis development of asthma.
Platelets can synthesize and release spasmogen, such as histamine, platelet activating factor (PAF), 5-HT, and thromboxane A2 (TxA2), that act on the airway smooth muscle cells to cause bronchoconstriction [75,76][51][52]. Depletion of platelets reduced allergen-induced airway hyperreactivity in allergic rabbits [77][53]. Studies on guinea pigs have shown that Bradykinin and capsaicin induced airways obstruction in a platelet-dependent manner [78][54]. PAF, a phospholipid derivative from various cells, including platelets, mast cells, basophils, eosinophils, and so on, is a strong activator of platelets and is well acknowledged for its role in bronchoconstriction, bronchial hyperresponsiveness, mucus hypersecretion, and gas exchange impairment [79][55]. TxA2, a potent airway smooth muscle contractile agent predominantly produced by platelets, is associated with airway inflammation and bronchial hyperreactivity [76][52]. Platelets act as a major reservoir and means of transport for brain-derived neurotrophic factor (BDNF). BDNF was found to contribute to airway obstruction and hyperresponsiveness in a model of allergic asthma [80][56]. Increased BDNF concentrations in platelets of asthmatic patients were found to correlate with the clinical parameters of airway dysfunction [32]. Recently, it was reported that the sphingolipid metabolism was altered in the patients allergic to house dust mites. Activated platelets are rich sources of sphingosine-1-phosphate (S1P), which acts directly on the smooth muscle cells to promote proliferation and AHR. The severity of allergen-induced bronchoconstriction is highly correlated with the plasma concentration of S1P [33].
Platelets are also rich in proteases, mitogens, and growth factors, which not only participate in the damage and repair process of allergic asthma, but also affect the phenotypes of airway epithelial cells, fibroblasts, and airway smooth muscle cells [81][57]. Metalloproteases (e.g., MMP2, MMP9), free radicals, and cationic proteins can degrade extracellular matrix, increase the vascular permeability of airway epithelia, and stimulate mucus secretion [60][58]. In murine models of chronic allergic inflammation, platelets are indispensable for the structural remodeling of the airway after chronic exposure to aerosolized allergens [23]. Platelet-derived growth factor (PDGF) promotes the proliferation of human airway smooth muscle cells as a mitogen, but is also involved in airway fibrosis and airway remodeling as a strong chemokine of fibroblasts [82,83,84][59][60][61]. In a mouse model with repeated exposure to allergens, PDGF overexpression and airway smooth muscle thickening were observed [85][62]. Vascular endothelial growth factor (VEGF) induces endothelial cell growth and angiogenesis and increases vascular permeability, and platelets secrete VEGF upon activation in vivo [86][63]. A study showed that VEGF levels are associated with the degree of vascularity and are inversely correlated with the airway caliber and level of AHR [87][64]. In allergic asthma, platelets inhibit eosinophil apoptosis by secreting granulocyte-macrophage colony-stimulating factor (GM-CSF), thus extending chronic inflammatory response and tissue damage [40]. In addition, GM-CSF enhances the 5-lipoxygenase (5-LO) activity in neutrophils and eosinophils and promotes the production of CysLTs, causing airway smooth muscle contraction, inflammatory cell recruitment, and tissue edema [68][65].
In conclusion, platelets interact with multiple inflammatory cells vital in the inflammatory process and development of allergic asthma occurs through direct cell-to-cell contact or inflammatory mediators, thus deeply involved in the pathogenesis of allergic asthma ( Figure 2 ).
4. Antiplatelet Therapies for Asthma Control
Currently, the treatments for allergic asthma include various strategies, including anti-inflammatory agents, bronchodilators, allergen-specific immunotherapy, and biologics targeting eosinophil activation and cytokines production, such as anti-IL-5 therapy. However, these strategies often do not result in full resolution for most asthmatic endotypes [88][66]. Hence, a new therapy to treat allergic asthma is urgently needed. Given that platelets are extensively involved in the pathogenesis of allergic asthma, it is possible in principle to treat allergic asthma with antiplatelet drugs ( Table 32 ). Common antiplatelet drugs include ADP receptor antagonists, thromboxane synthase inhibitors, thromboxane-prostanoid receptor (TP receptor) antagonists, 5-HT modifier, and cyclooxygenase (COX) inhibitors.
| Category | Target | Drug | Mechanism | References |
|---|---|---|---|---|
| ADP receptor antagonists | P2Y12 receptor | Clopidogrel Prasugrel Ticagrelor |
Reduce the release of platelets and the formation of platelets-leukocyte aggregates, and inhibit eosinophilic inflammation and airway hyperreactivity. | [67][68][69][70] |
| P2Y1 receptor | MRS2179 MRS2500 |
Inhibit the recruitment of eosinophils and lymphocytes to the lung. | [71] | |
| TxA2 synthase inhibitor | TxA2 synthase | Ozagrel (OKY-46) ONO-1301 |
Inhibit the production of proinflammatory cytokines and alleviate the eosinophil infiltration in the airways; suppress AHR and airway inflammation. | [72][73] |
| TP receptor antagonist | TP receptor | Seratrodast (AA-2414) S-1452 |
Reduce bronchial hyperresponsiveness by reducing airway inflammation. | [72][74] |
| 5-HT modifier | 5-HT-specific transporter | Tianeptine | Enhance the uptake of free 5-HT in peripheral blood. | [75][76] |
TxA2, a lipid metabolite produced by activated platelets through the arachidonic acid metabolic pathway, is an important proinflammatory mediator. In allergic asthma, TxA2 causes bronchial contraction, increased vascular permeability, tissue edema, and airway hyperresponsiveness [107][77]. Blocking TxA2 synthesis reduces the concentration of TxA2 during asthma attacks, but another bronchoconstrictor, prostaglandin, which acts together with TxA2 by binding to the TxA2 receptor (TP receptors), is generated due to the diverted arachidonic acid metabolism pathway [108][78]. Therefore, the combined use of TxA2 synthase inhibitor and TP receptor antagonist could be more effective in the treatment of allergic asthma. Indeed, the administration of Ozagrel (OKY-046, a selective TxA2 synthase inhibitor) and S-1452 (a TP receptor antagonist) inhibited the production of proinflammatory cytokines and prevented the infiltration of eosinophils into the airway [93][72]. Similarly, Seratrodast (AA-2414), a TP receptor antagonist, was reportedly able to reduce bronchial hyperresponsiveness by reducing airway inflammation [95][74]. ONO-1301, a novel prostacyclin agonist and TxA2 synthase inhibitor, was shown to suppress AHR and airway inflammation in asthma [94][73]. While TxA2 synthetase inhibitors and TP receptor antagonists were found to have positive effects on allergic asthma in Japan [109][79], studies from Western countries revealed no obvious curative effects. This discrepancy may be associated with the genetic polymorphism in TxA2 synthetase and TP receptors [108][78].
It is noteworthy that aspirin, a member of NSAIDs that is commonly used to prevent thrombosis, is a selective COX-1 inhibitor which plays an antithrombotic role by inhibiting TxA2 synthesis in platelets [114][80]. Clinical studies have revealed that some patients with allergic asthma show intolerance to aspirin and may even develop AERD, mainly due to the significantly reduced PGE 2 synthesis. Mast cell activation and the interaction between platelets and granulocytes lead to overexpression of CysLTs. All of these factors contribute to constriction of the bronchioles, acute exacerbation of asthma, and increased mucus production in AERD [115,116][81][82]. Therefore, aspirin should be used with caution when treating allergic asthma. Presently, AERD treatments include intravenous corticosteroids, anti-IgE, anti-IL-4/13, aspirin desensitization, and leukotriene-modifying drugs (leukotriene receptor antagonists and 5-lipooxygenase inhibitors) [70,71][83][84]. Due to the elevated levels of platelet activation and platelet-leukocyte aggregates in AERD, anti-platelet therapy would be an efficacious treatment, although clinical trials exploring the potential for platelet-targeted therapies in AERD are still in the early stages [11].
The application of antiplatelet drugs to allergic asthma needs to be further studied. Page CP pointed out in 1988 that platelets use different signaling pathways during hemostatic and inflammatory response. In an inflammatory response, inflammatory factors evoke platelet activation, and platelets participate in the inflammatory response through adhesion and secretion activities, but this generally does not include the aggregatory response [117][85]. Consistent with this notion, clinical studies found minor coagulation defects and prolonged bleeding time in patients with allergic asthma. Therefore, it is of great value to further study the underlying mechanism of platelets in the inflammatory process, which can provide a new direction for new therapeutics for allergic asthma.
5. New Insights of Platelets in Asthma
References
- Holgate, S.T.; Wenzel, S.; Postma, D.S.; Weiss, S.T.; Renz, H.; Sly, P.D. Asthma. Nat. Rev. Dis. Primers 2015, 1, 15025.
- Huang, K.; Yang, T.; Xu, J.; Yang, L.; Zhao, J.; Zhang, X.; Bai, C.; Kang, J.; Ran, P.; Shen, H.; et al. Prevalence, risk factors, and management of asthma in China: A national cross-sectional study. Lancet 2019, 394, 407–418.
- Radermecker, C.; Louis, R.; Bureau, F.; Marichal, T. Role of neutrophils in allergic asthma. Curr. Opin. Immunol. 2018, 54, 28–34.
- Kuo, C.S.; Pavlidis, S.; Loza, M.; Baribaud, F.; Rowe, A.; Pandis, I.; Hoda, U.; Rossios, C.; Sousa, A.; Wilson, S.J.; et al. A Transcriptome-driven Analysis of Epithelial Brushings and Bronchial Biopsies to Define Asthma Phenotypes in U-BIOPRED. Am. J. Respir. Crit. Care Med. 2017, 195, 443–455.
- Agache, I.; Rogozea, L. Endotypes in allergic diseases. Curr. Opin. Allergy Clin. Immunol. 2018, 18, 177–183.
- Thon, J.N.; Italiano, J.E. Platelets: Production, morphology and ultrastructure. In Antiplatelet Agents. Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2012; pp. 3–22.
- Pitchford, S.C. Defining a role for platelets in allergic inflammation. Biochem. Soc. Trans. 2007, 35, 1104–1108.
- Gremmel, T.; Frelinger, A.L., 3rd; Michelson, A.D. Platelet Physiology. Semin. Thromb. Hemost. 2016, 42, 191–204.
- Xu, X.R.; Zhang, D.; Oswald, B.E.; Carrim, N.; Wang, X.; Hou, Y.; Zhang, Q.; Lavalle, C.; McKeown, T.; Marshall, A.H.; et al. Platelets are versatile cells: New discoveries in hemostasis, thrombosis, immune responses, tumor metastasis and beyond. Crit. Rev. Clin. Lab. Sci. 2016, 53, 409–430.
- Tamagawa-Mineoka, R.; Katoh, N.; Ueda, E.; Masuda, K.; Kishimoto, S. Elevated platelet activation in patients with atopic dermatitis and psoriasis: Increased plasma levels of beta-thromboglobulin and platelet factor 4. Allergol. Int. 2008, 57, 391–396.
- Laidlaw, T.M.; Boyce, J.A. Platelets in patients with aspirin-exacerbated respiratory disease. J. Allergy Clin. Immunol. 2015, 135, 1407–1414.
- Idzko, M.; Pitchford, S.; Page, C. Role of platelets in allergic airway inflammation. J. Allergy Clin. Immunol. 2015, 135, 1416–1423.
- Morianos, I.; Semitekolou, M. Dendritic Cells: Critical Regulators of Allergic Asthma. Int. J. Mol. Sci. 2020, 21, 7930.
- Khan, D.A. Allergic rhinitis and asthma: Epidemiology and common pathophysiology. Allergy Asthma Proc. 2014, 35, 357–361.
- Schatz, M.; Rosenwasser, L. The allergic asthma phenotype. J. Allergy Clin. Immunology. Pract. 2014, 2, 645–648.
- Chen, S.; Deng, Y.; He, Q.; Chen, Y.; Wang, D.; Sun, W.; He, Y.; Zou, Z.; Liang, Z.; Chen, R.; et al. Toll-like Receptor 4 Deficiency Aggravates Airway Hyperresponsiveness and Inflammation by Impairing Neutrophil Apoptosis in a Toluene Diisocyanate-Induced Murine Asthma Model. Allergy Asthma Immunol. Res. 2020, 12, 608–625.
- Bazan-Socha, S.; Kuczia, P.; Potaczek, D.P.; Mastalerz, L.; Cybulska, A.; Zareba, L.; Kremers, R.; Hemker, C.; Undas, A. Increased blood levels of cellular fibronectin in asthma: Relation to the asthma severity, inflammation, and prothrombotic blood alterations. Respir. Med. 2018, 141, 64–71.
- De Boer, J.D.; Majoor, C.J.; Van ’t Veer, C.; Bel, E.H.; Van der Poll, T. Asthma and coagulation. Blood 2012, 119, 3236–3244.
- Nastałek, M.; Potaczek, D.P.; Wojas-Pelc, A.; Undas, A. Plasma platelet activation markers in patients with atopic dermatitis and concomitant allergic diseases. J. Dermatol. Sci. 2011, 64, 79–82.
- Kowal, K.; Pampuch, A.; Kowal-Bielecka, O.; DuBuske, L.M.; Bodzenta-Lukaszyk, A. Platelet activation in allergic asthma patients during allergen challenge with Dermatophagoides pteronyssinus. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2006, 36, 426–432.
- Metzger, W.J.; Sjoerdsma, K.; Richerson, H.B.; Moseley, P.; Zavala, D.; Monick, M.; Hunninghake, G.W. Platelets in bronchoalveolar lavage from asthmatic patients and allergic rabbits with allergen-induced late phase responses. Agents Actions Suppl. 1987, 21, 151–159.
- Averill, F.J.; Hubbard, W.C.; Proud, D.; Gleich, G.J.; Liu, M.C. Platelet activation in the lung after antigen challenge in a model of allergic asthma. Am. Rev. Respir. Dis 1992, 145, 571–576.
- Pitchford, S.C.; Riffo-Vasquez, Y.; Sousa, A.; Momi, S.; Gresele, P.; Spina, D.; Page, C.P. Platelets are necessary for airway wall remodeling in a murine model of chronic allergic inflammation. Blood 2004, 103, 639–647.
- Tutluoglu, B.; Gurel, C.B.; Ozdas, S.B.; Musellim, B.; Erturan, S.; Anakkaya, A.N.; Kilinc, G.; Ulutin, T. Platelet function and fibrinolytic activity in patients with bronchial asthma. Clin. Appl. Thromb. Hemost. 2005, 11, 77–81.
- Pitchford, S.C.; Yano, H.; Lever, R.; Riffo-Vasquez, Y.; Ciferri, S.; Rose, M.J.; Giannini, S.; Momi, S.; Spina, D.; O’Connor, B.; et al. Platelets are essential for leukocyte recruitment in allergic inflammation. J. Allergy Clin. Immunol. 2003, 112, 109–118.
- Kasperska-Zajac, A.; Brzoza, Z.; Rogala, B. Seasonal changes in platelet activity in pollen-induced seasonal allergic rhinitis and asthma. J. Asthma Off. J. Assoc. Care Asthma 2008, 45, 485–487.
- Benton, A.S.; Kumar, N.; Lerner, J.; Wiles, A.A.; Foerster, M.; Teach, S.J.; Freishtat, R.J. Airway platelet activation is associated with airway eosinophilic inflammation in asthma. J. Investig. Med. Off. Publ. Am. Fed. Clin. Res. 2010, 58, 987–990.
- Johansson, M.W.; Han, S.T.; Gunderson, K.A.; Busse, W.W.; Jarjour, N.N.; Mosher, D.F. Platelet activation, P-selectin, and eosinophil beta1-integrin activation in asthma. Am. J. Respir. Crit. Care Med. 2012, 185, 498–507.
- Johansson, M.W.; Mosher, D.F. Activation of beta1 integrins on blood eosinophils by P-selectin. Am. J. Respir. Cell Mol. Biol. 2011, 45, 889–897.
- Duarte, D.; Taveira-Gomes, T.; Sokhatska, O.; Palmares, C.; Costa, R.; Negrao, R.; Guimaraes, J.T.; Delgado, L.; Soares, R.; Moreira, A. Increased circulating platelet microparticles as a potential biomarker in asthma. Allergy 2013, 68, 1073–1075.
- Capron, M.; Jouault, T.; Prin, L.; Joseph, M.; Ameisen, J.C.; Butterworth, A.E.; Papin, J.P.; Kusnierz, J.P.; Capron, A. Functional study of a monoclonal antibody to IgE Fc receptor (Fc epsilon R2) of eosinophils, platelets, and macrophages. J. Exp. Med. 1986, 164, 72–89.
- Lommatzsch, M.; Schloetcke, K.; Klotz, J.; Schuhbaeck, K.; Zingler, D.; Zingler, C.; Schulte-Herbruggen, O.; Gill, H.; Schuff-Werner, P.; Virchow, J.C. Brain-derived neurotrophic factor in platelets and airflow limitation in asthma. Am. J. Respir. Crit. Care Med. 2005, 171, 115–120.
- Kowal, K.; Zebrowska, E.; Chabowski, A. Altered Sphingolipid Metabolism Is Associated With Asthma Phenotype in House Dust Mite-Allergic Patients. Allergy Asthma Immunol. Res. 2019, 11, 330–342.
- Hasegawa, S.; Pawankar, R.; Suzuki, K.; Nakahata, T.; Furukawa, S.; Okumura, K.; Ra, C. Functional expression of the high affinity receptor for IgE (FcepsilonRI) in human platelets and its’ intracellular expression in human megakaryocytes. Blood 1999, 93, 2543–2551.
- Hasegawa, S.; Tashiro, N.; Matsubara, T.; Furukawa, S.; Ra, C. A comparison of FcepsilonRI-mediated RANTES release from human platelets between allergic patients and healthy individuals. Int. Arch. Allergy Immunol. 2001, 125 (Suppl. 1), 42–47.
- Shah, S.A.; Kanabar, V.; Riffo-Vasquez, Y.; Mohamed, Z.; Cleary, S.J.; Corrigan, C.; James, A.L.; Elliot, J.G.; Shute, J.K.; Page, C.P.; et al. Platelets Independently Recruit into Asthmatic Lungs and Models of Allergic Inflammation via CCR3. Am. J. Respir. Cell Mol. Biol. 2021, 64, 557–568.
- Joseph, M.; Gounni, A.S.; Kusnierz, J.P.; Vorng, H.; Sarfati, M.; Kinet, J.P.; Tonnel, A.B.; Capron, A.; Capron, M. Expression and functions of the high-affinity IgE receptor on human platelets and megakaryocyte precursors. Eur. J. Immunol. 1997, 27, 2212–2218.
- Pitchford, S.C.; Momi, S.; Baglioni, S.; Casali, L.; Giannini, S.; Rossi, R.; Page, C.P.; Gresele, P. Allergen induces the migration of platelets to lung tissue in allergic asthma. Am. J. Respir. Crit. Care Med. 2008, 177, 604–612.
- Cardot, E.; Pestel, J.; Callebaut, I.; Lassalle, P.; Tsicopoulos, A.; Gras-Masse, H.; Capron, A.; Joseph, M. Specific activation of platelets from patients allergic to Dermatophagoides pteronyssinus by synthetic peptides derived from the allergen Der p I. Int. Arch. Allergy Immunol. 1992, 98, 127–134.
- Raiden, S.; Schettini, J.; Salamone, G.; Trevani, A.; Vermeulen, M.; Gamberale, R.; Giordano, M.; Geffner, J. Human platelets produce granulocyte-macrophage colony-stimulating factor and delay eosinophil apoptosis. Lab. Investig. J. Tech. Methods Pathol. 2003, 83, 589–598.
- Tsuji, T.; Nagata, K.; Koike, J.; Todoroki, N.; Irimura, T. Induction of superoxide anion production from monocytes an neutrophils by activated platelets through the P-selectin-sialyl Lewis X interaction. J. Leukoc. Biol. 1994, 56, 583–587.
- Nakanishi, T.; Inaba, M.; Inagaki-Katashiba, N.; Tanaka, A.; Vien, P.T.; Kibata, K.; Ito, T.; Nomura, S. Platelet-derived RANK ligand enhances CCL17 secretion from dendritic cells mediated by thymic stromal lymphopoietin. Platelets 2015, 26, 425–431.
- Pitchford, S.C.; Momi, S.; Giannini, S.; Casali, L.; Spina, D.; Page, C.P.; Gresele, P. Platelet P-selectin is required for pulmonary eosinophil and lymphocyte recruitment in a murine model of allergic inflammation. Blood 2005, 105, 2074–2081.
- Lukacs, N.W.; John, A.; Berlin, A.; Bullard, D.C.; Knibbs, R.; Stoolman, L.M. E- and P-selectins are essential for the development of cockroach allergen-induced airway responses. J. Immunol. (Baltimore, Md.: 1950) 2002, 169, 2120–2125.
- Cardenas, E.I.; Breaux, K.; Da, Q.; Flores, J.R.; Ramos, M.A.; Tuvim, M.J.; Burns, A.R.; Rumbaut, R.E.; Adachi, R. Platelet Munc13-4 regulates hemostasis, thrombosis and airway inflammation. Haematologica 2018, 103, 1235–1244.
- Yoshida, A.; Ohba, M.; Wu, X.; Sasano, T.; Nakamura, M.; Endo, Y. Accumulation of platelets in the lung and liver and their degranulation following antigen-challenge in sensitized mice. Br. J. Pharmacol. 2002, 137, 146–152.
- Kasperska-Zajac, A.; Rogala, B. Markers of platelet activation in plasma of patients suffering from persistent allergic rhinitis with or without asthma symptoms. Clin. Exp. Allergy 2005, 35, 1462–1465.
- Kasperska-Zajac, A.; Rogala, B. Platelet activity measured by plasma levels of beta-thromboglobulin and platelet factor 4 in seasonal allergic rhinitis during natural pollen exposure. Inflamm. Res. 2003, 52, 477–479.
- Koczy-Baron, E.; Grzanka, A.; Jochem, J.; Gawlik, R.; Kasperska-Zajac, A. Evaluation of circulating vascular endothelial growth factor and its soluble receptors in patients suffering from persistent allergic rhinitis. Allergy Asthma Clin. Immunol. Off. J. Can. Soc. Allergy Clin. Immunol. 2016, 12, 17.
- Potaczek, D.P. Links between allergy and cardiovascular or hemostatic system. Int. J. Cardiol. 2014, 170, 278–285.
- Herd, C.M.; Page, C.P. Pulmonary immune cells in health and disease: Platelets. Eur. Respir. J. 1994, 7, 1145–1160.
- Yoshimi, Y.; Fujimura, M.; Myou, S.; Tachibana, H.; Hirose, T. Effect of thromboxane A2 (TXA2) synthase inhibitor and TXA2 receptor antagonist alone and in combination on antigen-induced bronchoconstriction in guinea pigs. Prostaglandins Other Lipid Mediat. 2001, 65, 1–9.
- Coyle, A.J.; Page, C.P.; Atkinson, L.; Flanagan, R.; Metzger, W.J. The requirement for platelets in allergen-induced late asthmatic airway obstruction. Eosinophil infiltration and heightened airway responsiveness in allergic rabbits. Am. Rev. Respir. Dis. 1990, 142, 587–593.
- Keir, S.D.; Spina, D.; Page, C.P. Bradykinin and capsaicin induced airways obstruction in the guinea pig are platelet dependent. Pulm. Pharmacol. Ther. 2015, 33, 25–31.
- Kasperska-Zajac, A.; Brzoza, Z.; Rogala, B. Platelet activating factor as a mediator and therapeutic approach in bronchial asthma. Inflammation 2008, 31, 112–120.
- Braun, A.; Lommatzsch, M.; Neuhaus-Steinmetz, U.; Quarcoo, D.; Glaab, T.; McGregor, G.P.; Fischer, A.; Renz, H. Brain-derived neurotrophic factor (BDNF) contributes to neuronal dysfunction in a model of allergic airway inflammation. Br. J. Pharmacol. 2004, 141, 431–440.
- Barnes, P.J. New aspects of asthma. J. Intern. Med. 1992, 231, 453–461.
- Gomez-Casado, C.; Villasenor, A.; Rodriguez-Nogales, A.; Bueno, J.L.; Barber, D.; Escribese, M.M. Understanding Platelets in Infectious and Allergic Lung Diseases. Int. J. Mol. Sci. 2019, 20, 1730.
- Zhang, X.Y.; Tang, X.Y.; Li, N.; Zhao, L.M.; Guo, Y.L.; Li, X.S.; Tian, C.J.; Cheng, D.J.; Chen, Z.C.; Zhang, L.X. GAS5 promotes airway smooth muscle cell proliferation in asthma via controlling miR-10a/BDNF signaling pathway. Life Sci. 2018, 212, 93–101.
- Johnson, J.R.; Folestad, E.; Rowley, J.E.; Noll, E.M.; Walker, S.A.; Lloyd, C.M.; Rankin, S.M.; Pietras, K.; Eriksson, U.; Fuxe, J. Pericytes contribute to airway remodeling in a mouse model of chronic allergic asthma. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2015, 308, L658–L671.
- Wang, L.; Feng, X.; Hu, B.; Xia, Q.; Ni, X.; Song, Y. P2X4R promotes airway remodeling by acting on the phenotype switching of bronchial smooth muscle cells in rats. Purinergic Signal. 2018, 14, 433–442.
- Yamashita, N.; Sekine, K.; Miyasaka, T.; Kawashima, R.; Nakajima, Y.; Nakano, J.; Yamamoto, T.; Horiuchi, T.; Hirai, K.; Ohta, K. Platelet-derived growth factor is involved in the augmentation of airway responsiveness through remodeling of airways in diesel exhaust particulate-treated mice. J. Allergy Clin. Immunol. 2001, 107, 135–142.
- Wynendaele, W.; Derua, R.; Hoylaerts, M.F.; Pawinski, A.; Waelkens, E.; De Bruijn, E.A.; Paridaens, R.; Merlevede, W.; Van Oosterom, A.T. Vascular endothelial growth factor measured in platelet poor plasma allows optimal separation between cancer patients and volunteers: A key to study an angiogenic marker in vivo? Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 1999, 10, 965–971.
- Hur, G.Y.; Broide, D.H. Genes and Pathways Regulating Decline in Lung Function and Airway Remodeling in Asthma. Allergy Asthma Immunol. Res. 2019, 11, 604–621.
- Laidlaw, T.M.; Kidder, M.S.; Bhattacharyya, N.; Xing, W.; Shen, S.; Milne, G.L.; Castells, M.C.; Chhay, H.; Boyce, J.A. Cysteinyl leukotriene overproduction in aspirin-exacerbated respiratory disease is driven by platelet-adherent leukocytes. Blood 2012, 119, 3790–3798.
- Finotto, S. Resolution of allergic asthma. Semin. Immunopathol. 2019, 41, 665–674.
- Thomas, M.R.; Storey, R.F. Effect of P2Y12 inhibitors on inflammation and immunity. Thromb. Haemost. 2015, 114, 490–497.
- Storey, R.F.; Judge, H.M.; Wilcox, R.G.; Heptinstall, S. Inhibition of ADP-induced P-selectin expression and platelet-leukocyte conjugate formation by clopidogrel and the P2Y12 receptor antagonist AR-C69931MX but not aspirin. Thromb. Haemost. 2002, 88, 488–494.
- Suh, D.H.; Trinh, H.K.; Liu, J.N.; Pham le, D.; Park, S.M.; Park, H.S.; Shin, Y.S. P2Y12 antagonist attenuates eosinophilic inflammation and airway hyperresponsiveness in a mouse model of asthma. J. Cell. Mol. Med. 2016, 20, 333–341.
- Lussana, F.; Di Marco, F.; Terraneo, S.; Parati, M.; Razzari, C.; Scavone, M.; Femia, E.A.; Moro, A.; Centanni, S.; Cattaneo, M. Effect of prasugrel in patients with asthma: Results of PRINA, a randomized, double-blind, placebo-controlled, cross-over study. J. Thromb. Haemost. JTH 2015, 13, 136–141.
- Amison, R.T.; Momi, S.; Morris, A.; Manni, G.; Keir, S.; Gresele, P.; Page, C.P.; Pitchford, S.C. RhoA signaling through platelet P2Y(1) receptor controls leukocyte recruitment in allergic mice. J. Allergy Clin. Immunol. 2015, 135, 528–538.
- Shi, H.; Yokoyama, A.; Kohno, N.; Hirasawa, Y.; Kondo, K.; Sakai, K.; Hiwada, K. Effect of thromboxane A2 inhibitors on allergic pulmonary inflammation in mice. Eur. Respir. J. 1998, 11, 624–629.
- Hayashi, M.; Koya, T.; Kawakami, H.; Sakagami, T.; Hasegawa, T.; Kagamu, H.; Takada, T.; Sakai, Y.; Suzuki, E.; Gelfand, E.W.; et al. A prostacyclin agonist with thromboxane inhibitory activity for airway allergic inflammation in mice. Clin. Exp. Allergy J. Br. Soc. Allergy Clin. Immunol. 2010, 40, 317–326.
- Fukuoka, T.; Miyake, S.; Umino, T.; Inase, N.; Tojo, N.; Yoshizawa, Y. The effect of seratrodast on eosinophil cationic protein and symptoms in asthmatics. J. Asthma Off. J. Assoc. Care Asthma 2003, 40, 257–264.
- Chamba, G.; Lemoine, P.; Flachaire, E.; Ferry, N.; Quincy, C.; Sassard, J.; Ferber, C.; Mocaer, E.; Kamoun, A.; Renaud, B. Increased serotonin platelet uptake after tianeptine administration in depressed patients. Biol. Psychiatry 1991, 30, 609–617.
- Lechin, F.; Van der Dijs, B.; Lechin, A.E. Treatment of bronchial asthma with tianeptine. Methods Find. Exp. Clin. Pharmacol. 2004, 26, 697–701.
- Dogne, J.M.; De Leval, X.; Benoit, P.; Rolin, S.; Pirotte, B.; Masereel, B. Therapeutic potential of thromboxane inhibitors in asthma. Expert Opin. Investig. Drugs 2002, 11, 275–281.
- Hernandez, J.M.; Janssen, L.J. Revisiting the usefulness of thromboxane-A2 modulation in the treatment of bronchoconstriction in asthma. Can. J. Physiol. Pharm. 2015, 93, 111–117.
- Fujimura, M.; Sasaki, F.; Nakatsumi, Y.; Takahashi, Y.; Hifumi, S.; Taga, K.; Mifune, J.; Tanaka, T.; Matsuda, T. Effects of a thromboxane synthetase inhibitor (OKY-046) and a lipoxygenase inhibitor (AA-861) on bronchial responsiveness to acetylcholine in asthmatic subjects. Thorax 1986, 41, 955–959.
- Badimon, L.; Vilahur, G.; Rocca, B.; Patrono, C. The Key Contribution of Platelet and Vascular Arachidonic Acid Metabolism To The Pathophysiology Of Atherothrombosis. Cardiovasc. Res. 2021.
- Narayanankutty, A.; Resendiz-Hernandez, J.M.; Falfan-Valencia, R.; Teran, L.M. Biochemical pathogenesis of aspirin exacerbated respiratory disease (AERD). Clin. Biochem. 2013, 46, 566–578.
- Le Pham, D.; Lee, J.H.; Park, H.S. Aspirin-exacerbated respiratory disease: An update. Curr. Opin. Pulm. Med. 2017, 23, 89–96.
- Taniguchi, M.; Mitsui, C.; Hayashi, H.; Ono, E.; Kajiwara, K.; Mita, H.; Watai, K.; Kamide, Y.; Fukutomi, Y.; Sekiya, K.; et al. Aspirin-exacerbated respiratory disease (AERD): Current understanding of AERD. Allergol. Int. 2019, 68, 289–295.
- Dominas, C.; Gadkaree, S.; Maxfield, A.Z.; Gray, S.T.; Bergmark, R.W. Aspirin-exacerbated respiratory disease: A review. Laryngoscope Investig. Otolaryngol. 2020, 5, 360–367.
- Page, C.P. The involvement of platelets in non-thrombotic processes. Trends Pharmacol. Sci. 1988, 9, 66–71.
- Laumonnier, Y.; Wiese, A.V.; Figge, J.; Karsten, C. Regulation and function of anaphylatoxins and their receptors in allergic asthma. Mol. Immunol. 2017, 84, 51–56.
- Polley, M.J.; Nachman, R.L. Human platelet activation by C3a and C3a des-arg. J. Exp. Med. 1983, 158, 603–615.
- Sauter, R.J.; Sauter, M.; Obrich, M.; Emschermann, F.N.; Nording, H.; Patzelt, J.; Wendel, H.P.; Reil, J.C.; Edlich, F.; Langer, H.F. Anaphylatoxin Receptor C3aR Contributes to Platelet Function, Thrombus Formation and In Vivo Haemostasis. Thromb. Haemost. 2019, 119, 179–182.
- Subramaniam, S.; Jurk, K.; Hobohm, L.; Jäckel, S.; Saffarzadeplatelet activation and fibrin formatioh, M.; Schwierczek, K.; Wenzel, P.; Langer, F.; Reinhardt, C.; Ruf, W. Distinct contributions of complement factors to n in venous thrombus development. Blood 2017, 129, 2291–2302.
- Martel, C.; Cointe, S.; Maurice, P.; Matar, S.; Ghitescu, M.; Théroux, P.; Bonnefoy, A. Requirements for membrane attack complex formation and anaphylatoxins binding to collagen-activated platelets. PLoS ONE 2011, 6, e18812.
- Saradna, A.; Do, D.C.; Kumar, S.; Fu, Q.L.; Gao, P. Macrophage polarization and allergic asthma. Transl. Res. J. Lab. Clin. Med. 2018, 191, 1–14.
- Jiang, Z.; Zhu, L. Update on the role of alternatively activated macrophages in asthma. J. Asthma Allergy 2016, 9, 101–107.
- Rossaint, J.; Thomas, K.; Mersmann, S.; Skupski, J.; Margraf, A.; Tekath, T.; Jouvene, C.C.; Dalli, J.; Hidalgo, A.; Meuth, S.G.; et al. Platelets orchestrate the resolution of pulmonary inflammation in mice by T reg cell repositioning and macrophage education. J. Exp. Med. 2021, 218.
- Winnica, D.; Corey, C.; Mullett, S.; Reynolds, M.; Hill, G.; Wendell, S.; Que, L.; Holguin, F.; Shiva, S. Bioenergetic Differences in the Airway Epithelium of Lean Versus Obese Asthmatics Are Driven by Nitric Oxide and Reflected in Circulating Platelets. Antioxid. Redox Signal. 2019, 31, 673–686.
- Xu, W.; Cardenes, N.; Corey, C.; Erzurum, S.C.; Shiva, S. Platelets from Asthmatic Individuals Show Less Reliance on Glycolysis. PLoS ONE 2015, 10, e0132007.
- Newcomb, D.C.; Peebles, R.S., Jr. Th17-mediated inflammation in asthma. Curr. Opin. Immunol. 2013, 25, 755–760.
- Affandi, A.J.; Silva-Cardoso, S.C.; Garcia, S.; Leijten, E.F.A.; Van Kempen, T.S.; Marut, W.; Van Roon, J.A.G.; Radstake, T. CXCL4 is a novel inducer of human Th17 cells and correlates with IL-17 and IL-22 in psoriatic arthritis. Eur. J. Immunol. 2018, 48, 522–531.
- Ponomarev, E.D. Fresh Evidence for Platelets as Neuronal and Innate Immune Cells: Their Role in the Activation, Differentiation, and Deactivation of Th1, Th17, and Tregs during Tissue Inflammation. Front. Immunol 2018, 9, 406.
- Zhu, L.; Huang, Z.; Stalesen, R.; Hansson, G.K.; Li, N. Platelets provoke distinct dynamics of immune responses by differentially regulating CD4+ T-cell proliferation. J. Thromb. Haemost. JTH 2014, 12, 1156–1165.
- Weyrich, A.S.; Zimmerman, G.A. Platelets in lung biology. Annu. Rev. Physiol. 2013, 75, 569–591.
- Lefrançais, E.; Ortiz-Muñoz, G.; Caudrillier, A.; Mallavia, B.; Liu, F.; Sayah, D.M.; Thornton, E.E.; Headley, M.B.; David, T.; Coughlin, S.R.; et al. The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 2017, 544, 105–109.
- Yeung, A.K.; Villacorta-Martin, C.; Hon, S.; Rock, J.R.; Murphy, G.J. Lung megakaryocytes display distinct transcriptional and phenotypic properties. Blood Adv. 2020, 4, 6204–6217.
- Pariser, D.N.; Hilt, Z.T.; Ture, S.K.; Blick-Nitko, S.K.; Looney, M.R.; Cleary, S.J.; Roman-Pagan, E.; Saunders, J., 2nd; Georas, S.N.; Veazey, J.; et al. Lung megakaryocytes are immune modulatory cells. J. Clin. Investig. 2021, 131, e137377.
- Hogan, K.A.; Weiler, H.; Lord, S.T. Mouse models in coagulation. Thromb. Haemost. 2002, 87, 563–574.
- Alessandrini, F.; Musiol, S.; Schneider, E.; Blanco-Pérez, F.; Albrecht, M. Mimicking Antigen-Driven Asthma in Rodent Models-How Close Can We Get? Front. Immunol 2020, 11, 575936.
- Zschaler, J.; Schlorke, D.; Arnhold, J. Differences in innate immune response between man and mouse. Crit. Rev. Immunol. 2014, 34, 433–454.