The gut–brain axis is a bidirectional communication system driven by neural, hormonal, metabolic, immunological, and microbial signals. Signaling events from the gut can modulate brain function and recent evidence suggests that the gut–brain axis may play a pivotal role in linking gastrointestinal and neurological diseases. Accordingly, accumulating evidence has suggested a link between inflammatory bowel diseases (IBDs) and neurodegenerative, as well as neuroinflammatory diseases.
- gut-brain axis
- IBD
- MS
- PD
- ex vivo organ models
1. Introduction
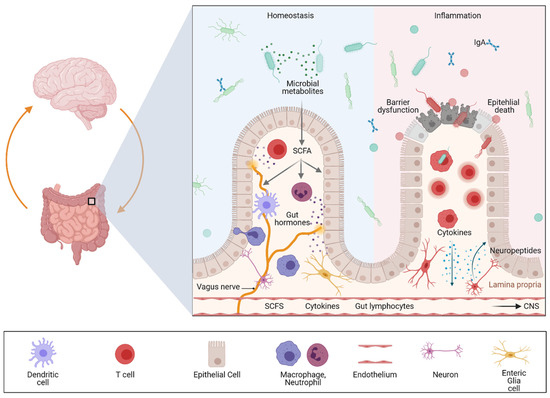
2. Genetic Evidence for an Association between Gut and Brain in the Context of Inflammation
The majority of PD cases are of sporadic origin; however, about 10% are familial and the most common monogenic forms of PD are pathogenic variants on the LRRK2 gene that encode leucine-rich repeat kinase 2 (LRRK2) [46][24], a multidomain protein with a catalytic core that can fulfill kinase and GTPase activity. It also has a scaffold function allowing LRRK2 to interact and recruit several other signaling molecules. The coding variants associated with PD cluster within the enzymatic core of LRRK2 and are thought to disrupt the enzymatic functions of this protein. Accordingly, preclinical studies have indicated that targeting the activity or expression of LRRK2 is neuroprotective.
Recent genome-wide association studies (GWAS) have shown that the association between the LRRK2 locus and IBD includes several LRRK2 genetic variants [49][25]. Although the interaction of alterations in LRRK2 function and CD mechanisms is still unknown, increasing evidence supports the fact that LRRK2 plays a role in mediating autophagy in Paneth cells, which would explain its strong association with CD because defects in Paneth cell autophagy have been described as hallmarks of this IBD prototype [52][26]. In line with this notion, preclinical studies using mice with a LRRK2 deficiency showed a specific impairment in the expression of antimicrobial peptides by Paneth cells [53][27].
The genetic risk factors associated with MS and IBD are not well described in the literature. Genetic studies of MS cohorts suggest that this autoimmune disease is provoked following exposure to environmental factors, which might be responsible for loss of tolerance and peripheral activation of myelin-specific T cells. GWAS have supported the complexity of MS pathology and uncovered immune-related gene variants linking MS to other autoimmune diseases such as IBD [64][28]. Further systematic studies are needed to better delineate the genetics between IBD and MS.
3. Evidence from Gut–Brain Communication in Preclinical Mouse Models
Beside the epidemiological and genetic evidence, several preclinical studies support the importance of gut–brain communication in intestinal inflammation. In the dextran sulfate sodium (DSS)-induced colitis, a rodent model of IBD, it was demonstrated that, parallel to local inflammatory responses in the gut mucosa, increased expression of IL6 and iNOS (Nitric oxide synthase, inducible; NOS2) was found in the cerebral cortex. The authors further described microglial activation by increased immunoreactivity for the pan-myeloid cell marker ionized calcium-binding adapter molecule 1 (Iba1) and elevated cytokine levels [65][29]. Another study analyzed the impact of intestinal inflammation by DSS administration in a model of dopaminergic neurodegeneration by LPS injection in the substantia nigra [66][30]. The authors demonstrated that inflammatory responses in the gut reinforced the inflammatory and deleterious effects of LPS induced neuroinflammation as indicated by increased levels of TNF-α, GFAP, and IL-6 in serum and the substantia nigra of the animals.
Interestingly, there are several recent animal studies suggesting that α-synuclein, a key protein involved in PD pathology, accumulates not only in the brain but also in the gut. Surprisingly, this could not only be observed in α-synuclein transgenic mice but also in mice subjected to DSS (experimental colitis drives enteric α-synuclein accumulation and Parkinson-like brain pathology). In line with previous examinations, this study observed that experimentally induced colitis in transgenic mice exacerbated α-synuclein pathologies in the CNS. While highly interesting, the effect of α-synuclein aggregates on ENS homeostasis has not been studied. So far, only one study demonstrated that increased α-synuclein expression following colitis was associated with phosphorylation in the myenteric plexus of common marmosets [68][31].
In summary, available studies suggest that inflammation is associated with peripheral alterations affecting CNS homeostasis through factors accumulating in the gut or systemically. In line with this hypothesis, another recent study demonstrated activation of microglial cells and reduction in occludin and claudin-5 expression in the brain suggesting an impaired BBB following experimental colitis [69][32]. These data further suggest that DSS-induced colitis increases systemic inflammation which then results in cortical inflammation via up-regulation of serum cytokines. Interestingly, the same group further observed that a decrease in dopaminergic function was associated with an increase in gastrointestinal inflammation, suggesting a bidirectional gut–brain interaction. Accordingly, mice studies showed that Experimental Autoimmune Encephalomyelitis (EAE), a model for MS in rodents, is accompanied by loss of mucosal immune homeostasis [70][33].
4. The Different Levels of Gut–Brain Communication
4.1. Neuronal Communication
The GI tract is the only internal organ that has its own independent nervous system, the enteric nervous system (ENS) [71,72][34][35]. This digestive system can be innervated by intrinsic enteric neurons and by extrinsic efferent and afferent nerves.
It has been shown that neuropeptide-containing (peptidergic) neurons within the colonic wall are key players in neurogenic inflammation as they release neuropeptides into the adjacent tissue. These peptides can induce vasodilation, plasma extravasation and leukocyte migration. Moreover, these neuropeptides have been shown to not only regulate intestinal homeostasis but also inflammation [76][36]. Accordingly, experimental studies could demonstrate that peptidergic neurons release neuropeptides that orchestrate colonic inflammation in a complex way. Calcitonin gene-related peptide (CGRP) and substance P (SP) seem to be the link between neuronal activation and the consecutive mucosal immune response. In murine colitis models, mice deficient in neutral endopeptidase (an enzyme responsible for the extracellular degradation of SP) displayed and aggravated colitis, while mice deficient in substance P showed a strong attenuation of colitis severity [15,74,77][15][37][38]. In sharp contrast, CGRP-deficient mice showed increased susceptibility to experimental colitis. Neuropeptide release is controlled by transient receptor potential (TRP) channels; therefore, neuropeptides released locally in the gut may function as mediators at the interface between the nervous system, the mucosal immune system and other cell compartments such as the epithelium or endothelium. Interestingly, recent single cell analyses revealed a significant expression of risk genes for diseases that feature intestinal and CNS involvement in the ENS, suggesting that it is involved in gut–brain disease communication [16].
4.2. Microbial Communication
While the importance of the gut microbiome was described some time ago for IBD and a variety of other immune and metabolically driven diseases, the essential role of the gut microbiota in CNS inflammation was discovered only a few years ago [78][39].
Several clinical studies highlighted a reduced diversity and altered composition of the gut microbiota (dysbiosis) not only in mouse models of neuroinflammation and neurodegeneration, but also as a common feature of patients with PD [23,80,81,82,83][40][41][42][43][44] and MS [84,85,86,87,88,89,90,91][45][46][47][48][49][50][51][52]. While dysbiosis has been shown in many clinical and preclinical studies, a disease-relevant microbiota for neuroinflammation or neurodegeneration is debatable. In addition, it still remains unclear whether dysbiosis can modulate inflammatory processes in the CNS or if it is merely the consequence of neuroinflammation/neurodegeneration. In support of a rather causative function of gut microbes, germ-free mice were resistant to spontaneous EAE, a striking notion that was explained by a lack of local activation of T cells in the gut and the subsequent deficient triggering of pathogenic antibody production by activated B cells. A translational study could demonstrate that transplanting faecal microbiota from PD patients exacerbated motor dysfunction in an α-synuclein transgenic mouse model [92][53].
4.3. Immunological Cross Talk
In summary, there is growing evidence suggesting that the gut is strongly involved in various neurological diseases via direct and indirect mechanisms. The key components are intestinal microbes and their products (e.g., metabolites) and immune education in the mucosal immune system, including immune cells releasing proinflammatory cytokines. Key to the regulation of these processes is the intestinal epithelium, which is capable of translating microbial and inflammatory signals to the immune system and secreting peptides as well as hormones, which are involved in the metabolic processing of dietary nutrients. Although this network is of strong clinical relevance for both intestinal and neurological diseases, we are just beginning to understand the underlying molecular mechanism and how organ crosstalk is regulated during health and disease. Accordingly, we need novel model systems to better understand microbiota–gut–brain communication on a cellular level. In the last chapter we therefore focus on novel human-specific preclinical model systems that will help to uncover disease mechanisms, which might allow us to better understand and modulate the function of this complex system.
5. Ex-Vivo Organ Models
5.1. Brain Organoids
5.2. Enteric Nervous System (ENS)
5.3. Intestinal Organoids
References
- Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342.
- Rogler, G.; Andus, T. Cytokines in inflammatory bowel disease. World J. Surg. 1998, 22, 382–389.
- Giraldez, M.D.; Carneros, D.; Garbers, C.; Rose-John, S.; Bustos, M. New insights into IL-6 family cytokines in metabolism, hepatology and gastroenterology. Nat. Rev. Gastroenterol. Hepatol. 2021, 5, 1–17.
- Khor, B.; Gardet, A.; Xavier, R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature 2011, 474, 307–317.
- Graham, D.B.; Xavier, R.J. Pathway paradigms revealed from the genetics of inflammatory bowel disease. Nature 2020, 578, 527–539.
- McGovern, D.P.; Kugathasan, S.; Cho, J.H. Genetics of Inflammatory Bowel Diseases. Gastroenterology 2015, 149, 1163–1176.e2.
- Gunther, C.; Martini, E.; Wittkopf, N.; Amann, K.; Weigmann, B.; Neumann, H.; Waldner, J.M.; Hedrick, S.M.; Tenzer, S.; Neurath, M.F.; et al. Caspase-8 regulates TNF-alpha-induced epithelial necroptosis and terminal ileitis. Nature 2011, 477, 335–339.
- Nenci, A.; Becker, C.; Wullaert, A.; Gareus, R.; van Loo, G.; Danese, S.; Huth, M.; Nikolaev, A.; Neufert, C.; Madison, B.; et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature 2007, 446, 557–561.
- Pastorelli, L.; De Salvo, C.; Mercado, J.R.; Vecchi, M.; Pizarro, T.T. Central role of the gut epithelial barrier in the pathogenesis of chronic intestinal inflammation: Lessons learned from animal models and human genetics. Front. Immunol. 2013, 4, 280.
- Miyoshi, J.; Sofia, M.A.; Pierre, J.F. The evidence for fungus in Crohn’s disease pathogenesis. Clin. J. Gastroenterol. 2018, 11, 449–456.
- Lee, M.; Chang, E.B. Inflammatory Bowel Diseases (IBD) and the Microbiome-Searching the Crime Scene for Clues. Gastroenterology 2021, 160, 524–537.
- Neurath, M.F. Host-microbiota interactions in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 76–77.
- Actis, G.C.; Ribaldone, D.G.; Pellicano, R. The Human Gut: Inflammatory Remote Manifestations Regulated by the Microbiome. J. Gastric Disord. Ther. 2019, 4, 24–27.
- Jarret, A.; Jackson, R.; Duizer, C.; Healy, M.E.; Zhao, J.; Rone, J.M.; Bielecki, P.; Sefik, E.; Roulis, M.; Rice, T.; et al. Enteric Nervous System-Derived IL-18 Orchestrates Mucosal Barrier Immunity. Cell 2020, 180, 50–63.e12.
- Engel, M.A.; Leffler, A.; Niedermirtl, F.; Babes, A.; Zimmermann, K.; Filipovic, M.R.; Izydorczyk, I.; Eberhardt, M.; Kichko, T.I.; Mueller-Tribbensee, S.M.; et al. TRPA1 and substance P mediate colitis in mice. Gastroenterology 2011, 141, 1346–1358.
- Drokhlyansky, E.; Smillie, C.S.; Van Wittenberghe, N.; Ericsson, M.; Griffin, G.K.; Eraslan, G.; Dionne, D.; Cuoco, M.S.; Goder-Reiser, M.N.; Sharova, T.; et al. The Human and Mouse Enteric Nervous System at Single-Cell Resolution. Cell 2020, 182, 1606–1622.e23.
- Hess, A.; Roesch, J.; Saake, M.; Sergeeva, M.; Hirschmann, S.; Neumann, H.; Dorfler, A.; Neurath, M.F.; Atreya, R. Functional Brain Imaging Reveals Rapid Blockade of Abdominal Pain Response Upon Anti-TNF Therapy in Crohn’s Disease. Gastroenterology 2015, 149, 864–866.
- Warren, S.; Sommers, S.C. Pathogenesis of ulcerative colitis. Am. J. Pathol. 1949, 25, 657–679.
- Truelove, S.C.; Witts, L.J. Cortisone in ulcerative colitis; final report on a therapeutic trial. Br. Med. J. 1955, 2, 1041–1048.
- Truelove, S.C.; Witts, L.J. Cortisone in ulcerative colitis; preliminary report on a therapeutic trial. Br. Med. J. 1954, 2, 375–378.
- Ng, Q.X.; Soh, A.Y.S.; Loke, W.; Lim, D.Y.; Yeo, W.S. The role of inflammation in irritable bowel syndrome (IBS). J. Inflamm. Res. 2018, 11, 345–349.
- Stabler, S.P. Clinical practice. Vitamin B12 deficiency. N. Engl. J. Med. 2013, 368, 149–160.
- Kosmidou, M.; Katsanos, A.H.; Katsanos, K.H.; Kyritsis, A.P.; Tsivgoulis, G.; Christodoulou, D.; Giannopoulos, S. Multiple sclerosis and inflammatory bowel diseases: A systematic review and meta-analysis. J. Neurol. 2017, 264, 254–259.
- Tolosa, E.; Vila, M.; Klein, C.; Rascol, O. LRRK2 in Parkinson disease: Challenges of clinical trials. Nat. Rev. Neurol. 2020, 16, 97–107.
- Witoelar, A.; Jansen, I.E.; Wang, Y.; Desikan, R.S.; Gibbs, J.R.; Blauwendraat, C.; Thompson, W.K.; Hernandez, D.G.; Djurovic, S.; Schork, A.J.; et al. Genome-wide Pleiotropy Between Parkinson Disease and Autoimmune Diseases. JAMA Neurol. 2017, 74, 780–792.
- Yang, E.; Shen, J. The roles and functions of Paneth cells in Crohn’s disease: A critical review. Cell Prolif. 2021, 54, e12958.
- Zhang, Q.; Pan, Y.; Yan, R.; Zeng, B.; Wang, H.; Zhang, X.; Li, W.; Wei, H.; Liu, Z. Commensal bacteria direct selective cargo sorting to promote symbiosis. Nat. Immunol. 2015, 16, 918–926.
- Sawcer, S.; Hellenthal, G.; Pirinen, M.; Spencer, C.C.; Patsopoulos, N.A.; Moutsianas, L.; Dilthey, A.; Su, Z.; Freeman, C.; Hunt, S.E.; et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011, 476, 214–219.
- Dempsey, E.; Abautret-Daly, A.; Docherty, N.G.; Medina, C.; Harkin, A. Persistent central inflammation and region specific cellular activation accompany depression- and anxiety-like behaviours during the resolution phase of experimental colitis. Brain Behav. Immun. 2019, 80, 616–632.
- Villaran, R.F.; Espinosa-Oliva, A.M.; Sarmiento, M.; De Pablos, R.M.; Arguelles, S.; Delgado-Cortes, M.J.; Sobrino, V.; Van Rooijen, N.; Venero, J.L.; Herrera, A.J.; et al. Ulcerative colitis exacerbates lipopolysaccharide-induced damage to the nigral dopaminergic system: Potential risk factor in Parkinson‘s disease. J. Neurochem. 2010, 114, 1687–1700.
- Resnikoff, H.; Metzger, J.M.; Lopez, M.; Bondarenko, V.; Mejia, A.; Simmons, H.A.; Emborg, M.E. Colonic inflammation affects myenteric alpha-synuclein in nonhuman primates. J. Inflamm. Res. 2019, 12, 113–126.
- Han, Y.; Zhao, T.; Cheng, X.; Zhao, M.; Gong, S.H.; Zhao, Y.Q.; Wu, H.T.; Fan, M.; Zhu, L.L. Cortical Inflammation is Increased in a DSS-Induced Colitis Mouse Model. Neurosci. Bull. 2018, 34, 1058–1066.
- Nouri, M.; Bredberg, A.; Westrom, B.; Lavasani, S. Intestinal barrier dysfunction develops at the onset of experimental autoimmune encephalomyelitis, and can be induced by adoptive transfer of auto-reactive T cells. PLoS ONE 2014, 9, e106335.
- Kunze, W.A.; Bornstein, J.C.; Furness, J.B. Identification of sensory nerve cells in a peripheral organ (the intestine) of a mammal. Neuroscience 1995, 66, 1–4.
- Spencer, N.J.; Hu, H. Enteric nervous system: Sensory transduction, neural circuits and gastrointestinal motility. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 338–351.
- Engel, M.A.; Becker, C.; Reeh, P.W.; Neurath, M.F. Role of sensory neurons in colitis: Increasing evidence for a neuroimmune link in the gut. Inflamm. Bowel. Dis. 2011, 17, 1030–1033.
- Di Giovangiulio, M.; Verheijden, S.; Bosmans, G.; Stakenborg, N.; Boeckxstaens, G.E.; Matteoli, G. The Neuromodulation of the Intestinal Immune System and Its Relevance in Inflammatory Bowel Disease. Front. Immunol. 2015, 6, 590.
- Engel, M.A.; Khalil, M.; Mueller-Tribbensee, S.M.; Becker, C.; Neuhuber, W.L.; Neurath, M.F.; Reeh, P.W. The proximodistal aggravation of colitis depends on substance P released from TRPV1-expressing sensory neurons. J. Gastroenterol. 2012, 47, 256–265.
- Berer, K.; Mues, M.; Koutrolos, M.; Rasbi, Z.A.; Boziki, M.; Johner, C.; Wekerle, H.; Krishnamoorthy, G. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature 2011, 479, 538–541.
- Cosma-Grigorov, A.; Meixner, H.; Mrochen, A.; Wirtz, S.; Winkler, J.; Marxreiter, F. Changes in Gastrointestinal Microbiome Composition in PD: A Pivotal Role of Covariates. Front. Neurol. 2020, 11, 1041.
- Boertien, J.M.; Pereira, P.A.B.; Aho, V.T.E.; Scheperjans, F. Increasing Comparability and Utility of Gut Microbiome Studies in Parkinson’s Disease: A Systematic Review. J. Parkinsons. Dis. 2019, 9, S297–S312.
- Barichella, M.; Severgnini, M.; Cilia, R.; Cassani, E.; Bolliri, C.; Caronni, S.; Ferri, V.; Cancello, R.; Ceccarani, C.; Faierman, S.; et al. Unraveling gut microbiota in Parkinson’s disease and atypical parkinsonism. Mov. Disord. 2019, 34, 396–405.
- Heintz-Buschart, A.; Pandey, U.; Wicke, T.; Sixel-Doring, F.; Janzen, A.; Sittig-Wiegand, E.; Trenkwalder, C.; Oertel, W.H.; Mollenhauer, B.; Wilmes, P. The nasal and gut microbiome in Parkinson’s disease and idiopathic rapid eye movement sleep behavior disorder. Mov. Disord. 2018, 33, 88–98.
- Lubomski, M.; Tan, A.H.; Lim, S.Y.; Holmes, A.J.; Davis, R.L.; Sue, C.M. Parkinson’s disease and the gastrointestinal microbiome. J. Neurol. 2020, 267, 2507–2523.
- Miyake, S.; Kim, S.; Suda, W.; Kawasumi, M.; Onawa, S.; Taguchi-Atarashi, N.; Morita, H.; Taylor, T.D.; Hattori, M.; Ohno, H. Dysbiosis in the Gut Microbiota of Patients with Multiple Sclerosis, with a Striking Depletion of Species Belonging to Clostridia XIVa and IV Clusters. PLoS ONE 2015, 10, e0137429.
- Tremlett, H.; Fadrosh, D.W.; Faruqi, A.A.; Hart, J.; Roalstad, S.; Graves, J.; Spencer, C.M.; Lynch, S.V.; Zamvil, S.S.; Waubant, E.; et al. Gut microbiota in early pediatric multiple sclerosis: A case-control study. Eur. J. Neurol. 2016, 23, 1308–1321.
- Jangi, S.; Gandhi, R.; Cox, L.M.; Li, N.; von Glehn, F.; Yan, R.; Patel, B.; Mazzola, M.A.; Liu, S.; Glanz, B.L.; et al. Alterations of the human gut microbiome in multiple sclerosis. Nat. Commun. 2016, 7, 12015.
- Chen, J.; Chia, N.; Kalari, K.R.; Yao, J.Z.; Novotna, M.; Paz Soldan, M.M.; Luckey, D.H.; Marietta, E.V.; Jeraldo, P.R.; Chen, X.; et al. Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Sci. Rep. 2016, 6, 28484.
- Berer, K.; Gerdes, L.A.; Cekanaviciute, E.; Jia, X.; Xiao, L.; Xia, Z.; Liu, C.; Klotz, L.; Stauffer, U.; Baranzini, S.E.; et al. Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 10719–10724.
- Cekanaviciute, E.; Yoo, B.B.; Runia, T.F.; Debelius, J.W.; Singh, S.; Nelson, C.A.; Kanner, R.; Bencosme, Y.; Lee, Y.K.; Hauser, S.L.; et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc. Natl. Acad. Sci. USA 2017, 114, 10713–10718.
- Montgomery, T.L.; Kunstner, A.; Kennedy, J.J.; Fang, Q.; Asarian, L.; Culp-Hill, R.; D’Alessandro, A.; Teuscher, C.; Busch, H.; Krementsov, D.N. Interactions between host genetics and gut microbiota determine susceptibility to CNS autoimmunity. Proc. Natl. Acad. Sci. USA 2020, 117, 27516–27527.
- Tremlett, H.; Fadrosh, D.W.; Faruqi, A.A.; Hart, J.; Roalstad, S.; Graves, J.; Spencer, C.M.; Lynch, S.V.; Zamvil, S.S.; Waubant, E.; et al. Associations between the gut microbiota and host immune markers in pediatric multiple sclerosis and controls. BMC Neurol. 2016, 16, 182.
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480.e12.
- Zhang, S.C.; Wernig, M.; Duncan, I.D.; Brustle, O.; Thomson, J.A. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat. Biotechnol. 2001, 19, 1129–1133.
- Eiraku, M.; Watanabe, K.; Matsuo-Takasaki, M.; Kawada, M.; Yonemura, S.; Matsumura, M.; Wataya, T.; Nishiyama, A.; Muguruma, K.; Sasai, Y. Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Cell Stem. Cell 2008, 3, 519–532.
- Sasai, Y. Next-generation regenerative medicine: Organogenesis from stem cells in 3D culture. Cell Stem. Cell 2013, 12, 520–530.
- Mariani, J.; Simonini, M.V.; Palejev, D.; Tomasini, L.; Coppola, G.; Szekely, A.M.; Horvath, T.L.; Vaccarino, F.M. Modeling human cortical development in vitro using induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 12770–12775.
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379.
- Kadoshima, T.; Sakaguchi, H.; Nakano, T.; Soen, M.; Ando, S.; Eiraku, M.; Sasai, Y. Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proc. Natl. Acad. Sci. USA 2013, 110, 20284–202849.
- Camp, J.G.; Badsha, F.; Florio, M.; Kanton, S.; Gerber, T.; Wilsch-Brauninger, M.; Lewitus, E.; Sykes, A.; Hevers, W.; Lancaster, M.; et al. Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc. Natl. Acad. Sci. USA 2015, 112, 15672–15677.
- Pasca, A.M.; Sloan, S.A.; Clarke, L.E.; Tian, Y.; Makinson, C.D.; Huber, N.; Kim, C.H.; Park, J.Y.; O’Rourke, N.A.; Nguyen, K.D.; et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods 2015, 12, 671–678.
- Jo, J.; Xiao, Y.; Sun, A.X.; Cukuroglu, E.; Tran, H.D.; Goke, J.; Tan, Z.Y.; Saw, T.Y.; Tan, C.P.; Lokman, H.; et al. Midbrain-like Organoids from Human Pluripotent Stem Cells Contain Functional Dopaminergic and Neuromelanin-Producing Neurons. Cell Stem. Cell 2016, 19, 248–257.
- Sakaguchi, H.; Kadoshima, T.; Soen, M.; Narii, N.; Ishida, Y.; Ohgushi, M.; Takahashi, J.; Eiraku, M.; Sasai, Y. Generation of functional hippocampal neurons from self-organizing human embryonic stem cell-derived dorsomedial telencephalic tissue. Nat. Commun. 2015, 6, 8896.
- Pellegrini, L.; Bonfio, C.; Chadwick, J.; Begum, F.; Skehel, M.; Lancaster, M.A. Human CNS barrier-forming organoids with cerebrospinal fluid production. Science 2020, 369, 6500.
- Birey, F.; Andersen, J.; Makinson, C.D.; Islam, S.; Wei, W.; Huber, N.; Fan, H.C.; Metzler, K.R.C.; Panagiotakos, G.; Thom, N.; et al. Assembly of functionally integrated human forebrain spheroids. Nature 2017, 545, 54–59.
- Bagley, J.A.; Reumann, D.; Bian, S.; Levi-Strauss, J.; Knoblich, J.A. Fused cerebral organoids model interactions between brain regions. Nat. Methods 2017, 14, 743–751.
- Andersen, J.; Revah, O.; Miura, Y.; Thom, N.; Amin, N.D.; Kelley, K.W.; Singh, M.; Chen, X.; Thete, M.V.; Walczak, E.M.; et al. Generation of Functional Human 3D Cortico-Motor Assembloids. Cell 2020, 183, 1913–1929.e26.
- Barber, K.; Studer, L.; Fattahi, F. Derivation of enteric neuron lineages from human pluripotent stem cells. Nat. Protoc. 2019, 14, 1261–1279.
- Fattahi, F.; Steinbeck, J.A.; Kriks, S.; Tchieu, J.; Zimmer, B.; Kishinevsky, S.; Zeltner, N.; Mica, Y.; El-Nachef, W.; Zhao, H.; et al. Deriving human ENS lineages for cell therapy and drug discovery in Hirschsprung disease. Nature 2016, 531, 105–109.
- Zorn, A.M.; Wells, J.M. Vertebrate endoderm development and organ formation. Annu. Rev. Cell Dev. Biol. 2009, 25, 221–251.
- Gao, S.; Yan, L.; Wang, R.; Li, J.; Yong, J.; Zhou, X.; Wei, Y.; Wu, X.; Wang, X.; Fan, X.; et al. Tracing the temporal-spatial transcriptome landscapes of the human fetal digestive tract using single-cell RNA-sequencing. Nat. Cell Biol. 2018, 20, 721–734.
- Lancaster, M.A.; Huch, M. Disease modelling in human organoids. Dis. Model. Mech. 2019, 12, 039347.
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130.
- Rowe, R.G.; Daley, G.Q. Induced pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Genet. 2019, 20, 377–388.
- Spence, J.R.; Mayhew, C.N.; Rankin, S.A.; Kuhar, M.F.; Vallance, J.E.; Tolle, K.; Hoskins, E.E.; Kalinichenko, V.V.; Wells, S.I.; Zorn, A.M.; et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 2011, 470, 105–109.
- Mithal, A.; Capilla, A.; Heinze, D.; Berical, A.; Villacorta-Martin, C.; Vedaie, M.; Jacob, A.; Abo, K.; Szymaniak, A.; Peasley, M.; et al. Generation of mesenchyme free intestinal organoids from human induced pluripotent stem cells. Nat. Commun. 2020, 11, 215.
- Forster, R.; Chiba, K.; Schaeffer, L.; Regalado, S.G.; Lai, C.S.; Gao, Q.; Kiani, S.; Farin, H.F.; Clevers, H.; Cost, G.J.; et al. Human intestinal tissue with adult stem cell properties derived from pluripotent stem cells. Stem. Cell Rep. 2014, 2, 838–852.
- McCracken, K.W.; Howell, J.C.; Wells, J.M.; Spence, J.R. Generating human intestinal tissue from pluripotent stem cells in vitro. Nat. Protoc. 2011, 6, 1920–1928.
- Workman, M.J.; Mahe, M.M.; Trisno, S.; Poling, H.M.; Watson, C.L.; Sundaram, N.; Chang, C.F.; Schiesser, J.; Aubert, P.; Stanley, E.G.; et al. Engineered human pluripotent-stem-cell-derived intestinal tissues with a functional enteric nervous system. Nat. Med. 2017, 23, 49–59.
- Sarvestani, S.K.; Signs, S.; Hu, B.; Yeu, Y.; Feng, H.; Ni, Y.; Hill, D.R.; Fisher, R.C.; Ferrandon, S.; DeHaan, R.K.; et al. Induced organoids derived from patients with ulcerative colitis recapitulate colitic reactivity. Nat. Commun. 2021, 12, 262.
- Yoshida, S.; Miwa, H.; Kawachi, T.; Kume, S.; Takahashi, K. Generation of intestinal organoids derived from human pluripotent stem cells for drug testing. Sci. Rep. 2020, 10, 5989.
- van der Flier, L.G.; Clevers, H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu. Rev. Physiol. 2009, 71, 241–260.
- Williams, J.M.; Duckworth, C.A.; Burkitt, M.D.; Watson, A.J.; Campbell, B.J.; Pritchard, D.M. Epithelial cell shedding and barrier function: A matter of life and death at the small intestinal villus tip. Vet. Pathol. 2015, 52, 445–455.
- Alison, M.R.; Lin, W.R. Hepatocyte turnover and regeneration: Virtually a virtuoso performance. Hepatology 2011, 53, 1393–1396.
- Kretzschmar, K.; Clevers, H. Organoids: Modeling Development and the Stem Cell Niche in a Dish. Dev. Cell 2016, 38, 590–600.
- Gunther, C.; Brevini, T.; Sampaziotis, F.; Neurath, M.F. What gastroenterologists and hepatologists should know about organoids in 2019. Dig. Liver Dis. 2019, 51, 753–760.
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265.
- Min, S.; Kim, S.; Cho, S.W. Gastrointestinal tract modeling using organoids engineered with cellular and microbiota niches. Exp. Mol. Med. 2020, 52, 227–237.
- Hefele, M.; Stolzer, I.; Ruder, B.; He, G.W.; Mahapatro, M.; Wirtz, S.; Neurath, M.F.; Gunther, C. Intestinal epithelial Caspase-8 signaling is essential to prevent necroptosis during Salmonella Typhimurium induced enteritis. Mucosal. Immunol. 2018, 11, 1191–1202.
- Gunther, C.; Ruder, B.; Stolzer, I.; Dorner, H.; He, G.W.; Chiriac, M.T.; Aden, K.; Strigli, A.; Bittel, M.; Zeissig, S.; et al. Interferon Lambda Promotes Paneth Cell Death Via STAT1 Signaling in Mice and Is Increased in Inflamed Ileal Tissues of Patients With Crohn’s Disease. Gastroenterology 2019, 157, 1310–1322.e13.
- Bittel, M.; Kremer, A.E.; Sturzl, M.; Wirtz, S.; Stolzer, I.; Neurath, M.F.; Ballon, G.; Gunther, C. Modulation of the extrinsic cell death signaling pathway by viral Flip induces acute-death mediated liver failure. Cell Death Dis. 2019, 10, 878.
- Bartfeld, S.; Bayram, T.; van de Wetering, M.; Huch, M.; Begthel, H.; Kujala, P.; Vries, R.; Peters, P.J.; Clevers, H. In vitro expansion of human gastric epithelial stem cells and their responses to bacterial infection. Gastroenterology 2015, 148, 126–136.e6.
- Boccellato, F.; Woelffling, S.; Imai-Matsushima, A.; Sanchez, G.; Goosmann, C.; Schmid, M.; Berger, H.; Morey, P.; Denecke, C.; Ordemann, J.; et al. Polarised epithelial monolayers of the gastric mucosa reveal insights into mucosal homeostasis and defence against infection. Gut 2019, 68, 400–413.
- Wroblewski, L.E.; Piazuelo, M.B.; Chaturvedi, R.; Schumacher, M.; Aihara, E.; Feng, R.; Noto, J.M.; Delgado, A.; Israel, D.A.; Zavros, Y.; et al. Helicobacter pylori targets cancer-associated apical-junctional constituents in gastroids and gastric epithelial cells. Gut 2015, 64, 720–730.
- Shaffiey, S.A.; Jia, H.; Keane, T.; Costello, C.; Wasserman, D.; Quidgley, M.; Dziki, J.; Badylak, S.; Sodhi, C.P.; March, J.C.; et al. Intestinal stem cell growth and differentiation on a tubular scaffold with evaluation in small and large animals. Regen. Med. 2016, 11, 45–61.
- Hou, Q.; Ye, L.; Liu, H.; Huang, L.; Yang, Q.; Turner, J.R.; Yu, Q. Correction: Lactobacillus accelerates ISCs regeneration to protect the integrity of intestinal mucosa through activation of STAT3 signaling pathway induced by LPLs secretion of IL-22. Cell Death Differ. 2021, 28, 2025–2027.
- VanDussen, K.L.; Marinshaw, J.M.; Shaikh, N.; Miyoshi, H.; Moon, C.; Tarr, P.I.; Ciorba, M.A.; Stappenbeck, T.S. Development of an enhanced human gastrointestinal epithelial culture system to facilitate patient-based assays. Gut 2015, 64, 911–920.