Liver sinusoids are lined by liver sinusoidal endothelial cells (LSEC), which represent approximately 15 to 20% of the liver cells, but only 3% of the total liver volume. LSEC have unique functions, such as fluid filtration, blood vessel tone modulation, blood clotting, inflammatory cell recruitment, and metabolite and hormone trafficking. Different subtypes of liver endothelial cells are also known to control liver zonation and hepatocyte function. The liver has the exceptional ability to regenerate from small remnants. The past decades have seen increasing awareness in the role of non-parenchymal cells in liver regeneration despite not being the most represented population. While a lot of knowledge has emerged, clarification is needed regarding the role of LSEC in sensing shear stress and on their participation in the inductive phase of regeneration by priming the hepatocytes and delivering mitogenic factors. It is also unclear if bone marrow-derived LSEC participate in the proliferative phase of liver regeneration. Similarly, data are scarce as to LSEC having a role in the termination phase of the regeneration process. Here, we review what is known about the interaction between LSEC and other liver cells during the different phases of liver regeneration. We next explain extended hepatectomy and small liver transplantation, which lead to “small for size syndrome” (SFSS), a lethal liver failure. SFSS is linked to endothelial denudation, necrosis, and lobular disturbance. Using the knowledge learned from partial hepatectomy studies on LSEC, we expose several techniques that are, or could be, used to avoid the “small for size syndrome” after extended hepatectomy or small liver transplantation.
- liver sinusoidal endothelial cells
- liver regeneration
- angiogenesis
- endothelial progenitor cell
- shear stress
- partial hepatectomy
- extended hepatectomy
- small for size syndrome
- post-hepatectomy liver failure
1. Introduction
The extraordinary ability of the liver to regenerate has been known since the Antiquity. The cellular and molecular mechanisms supporting regeneration have being intensely studied for decades. Yet, understanding how the process is fine-tuned to maintain an appropriate cell mass, cell composition and cell organization for an efficient function during lifetime homeostasis and wound healing remains a mystery. Hepatocytes, which accomplish numerous metabolic functions, represent 60% of all liver cells and account for 80% of the liver mass [1]. A large bulk of them, mainly midzonal hepatocytes [2], enter the replicative program when liver mass abruptly decreases as after toxic, ischemic, or viral insults or after surgical removal of part of the organ to restore volume and function. Kupffer cells (KC) and other hepatic immune cells, hepatic stellate cells (HSC), cholangiocytes, and LSEC interact with hepatocytes to support hepatocyte regeneration and ensure a functional structure of the lobule [3][4][5][6][7]. In this review, we will focus on how LSEC take part in each step of liver regeneration, from the inductive phase to the termination phase.
12. LSEC Renewal during Homeostasis
2
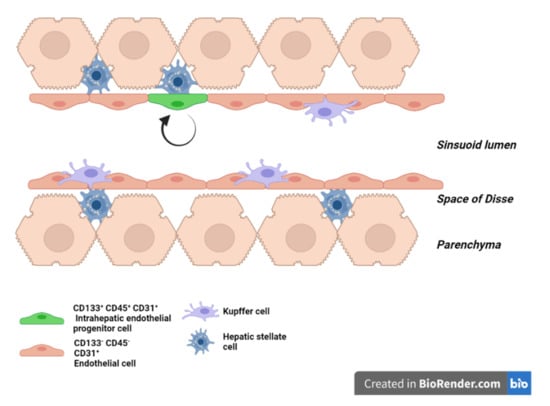
3. LSEC during Regeneration after Partial Hepatectomy
23.1. LSEC during the Inductive Phase of Liver Regeneration
23.1.1. How Do LSEC Modulate Hepatocyte Regeneration
[3][5][6][7]. Right after PHx, LSEC experience an increased shear stress due to the brutal redirection of the entire portal blood flow, normally distributed to the full-size
organ, into the smaller vascular bed of the liver remnant. The larger the resection, the larger the mismatch between the portal blood inflow and the remnant vascular network
[5]
[25]
[25]
[13]. An alternative and not mutually exclusive explanation of the endothelial shear-stress stimulus for regeneration is that the increased liver inflow (through the portal vein) brings larger amounts of growth factors to the liver remnant stimulating cell proliferation more
vigorously. Such growth factors come from the pancreas (insulin) or from the intestine (such as epithelial growth factor (EGF) produced by the duodenal Brunner’s glands)
[3][26][27][28]. A change in the exposure of the remnant liver and of the remnant sinusoidal bed to gut microbial products (including lipopolysaccharides) according to liver inflow might also contribute to
the modulation of the regenerative response
[29]
23.1.2. How Do LSEC Interact with Hepatocytes, NPC’s and Circulating Progenitors
[31]
[34]
[35]
[39]. Subsequently, Wang et al. proposed that HGF and Wnt2- rich bone marrow-derived endothelial progenitor cells are recruited to the regenerating liver
[9]
[43]
[9]
[44]
[9].
Moreover, the fact that the injection of bone marrow progenitors at day three did not rescue liver growth, propounded the idea that LSEC stimulate hepatocyte proliferation in early timings after PHx to coordinate liver regeneration
[49]
[54]. LSEC also interact with platelets and monocytes. After hepatectomy, platelets adhere to LSEC and activate them to secrete growth factors such as IL-6. These proteins stimulate proliferation of hepatocytes to ensure liver regeneration
[55]
[56]
[57]. Figure 2 recapitulates in a schematic manner the essential role of native LSEC and recruited bone marrow-derived LSEC as early initiators and coordinators for hepatocyte proliferation and liver regeneration. More research is needed, in order to analyze the different endothelial sub-populations and to study their respective spatio-temporal contribution to regeneration.
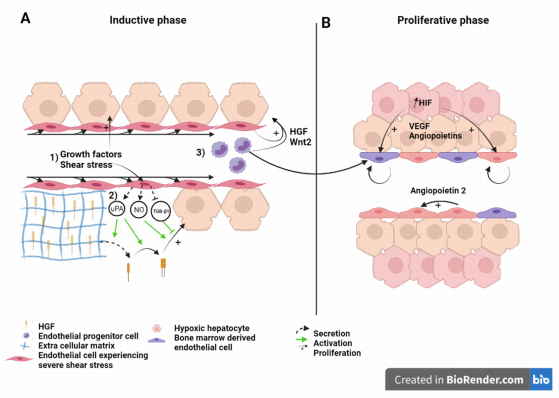
Figure 2. Liver sinusoid and endothelial cells during the (A) inductive phase and (B) angiogenic phase of liver regeneration
after partial hepatectomy. Scheme on the role of LSEC during regeneration after partial hepatectomy during the inductive
phase (A) where (1) growth factors, (2) increased shear stress, as well as (3) bone marrow endothelial progenitors induce
the proliferation of the hepatocytes. This is also overused during the proliferative phase (B), where LSEC upregulate
angiopoietin-2 paracrine secretion and proliferating hepatocytes, which experience a relative hypoxia, secrete pro-angiogenic
factors to induce the proliferation of LSEC. Recruited endothelial progenitor cells become LSEC during regeneration. HGF:
Hepatic growth factor; NO: Nitric oxide; TGB-β1: Tumor growth factor beta 1; Wnt2: Wingless-type MMTV integration site
family, member 2; HIF: Hypoxia inducible factor; VEGF: Vascular endothelial growth factor.
Liver sinusoid and endothelial cells during the (A) inductive phase and (B) angiogenic phase of liver regeneration after partial hepatectomy. Scheme on the role of LSEC during regeneration after partial hepatectomy during the inductive phase (A) where (1) growth factors, (2) increased shear stress, as well as (3) bone marrow endothelial progenitors induce the proliferation of the hepatocytes. This is also overused during the proliferative phase (B), where LSEC upregulate angiopoietin-2 paracrine secretion and proliferating hepatocytes, which experience a relative hypoxia, secrete pro-angiogenic factors to induce the proliferation of LSEC. Recruited endothelial progenitor cells become LSEC during regeneration. HGF: Hepatic growth factor; NO: Nitric oxide; TGB-β1: Tumor growth factor beta 1; Wnt2: Wingless-type MMTV integration site family, member 2; HIF: Hypoxia inducible factor; VEGF: Vascular endothelial growth factor.
23.2. LSEC during the Angiogenic Phase of Liver Regeneration
During the angiogenic phase, LSEC upregulates the expression of angiopoietin-2. This pro-angiogenic factor indirectly stimulates LSEC proliferation in a paracrine manner through upregulation of VEGFR2 [58] (Figure 2). VEGFR2 is the main mediator of VEGF signal during liver regeneration [8]. Hepatocytes engaged in cell cycle or that newly completed cell division also express pro-angiogenic factors, mainly VEGF and angiopoietins, that subsequently stimulate a pro-angiogenic response characterized by DNA synthesis and cell duplication of LSEC [59][60][61]. Hepatocytes, which number has increased upon cell division, experience relative hypoxia that engage the hypoxia-inducible factor (HIF) pathway and the downstream production of pro-angiogenic factors [62] (Figure 2B). Subsequently, endothelial cell proliferation leads to the elongation of the sinusoidal network. While hepatocyte replication reaches its maximum 24 to 48 h after hepatectomy in rats, and mice, respectively, LSEC proliferation peaks at post-surgery day 3 to 4 in rodents.
23.3. LSEC during the Termination Phase of Liver Regeneration
Overall, research has shown that LSEC and endothelial progenitors are essential in
liver regeneration initiation, proliferation and termination phases, and a decrease of bonemarrow-derived cells or/and a decrease of factors secreted by endothelial cells negatively impact the liver re-growth.
34. Role of LSEC in Extended Hepatectomy
Post-hepatectomy liver failure (PHLF) (or post liver transplantation “Small-for-size syndrome” (SFSS)) is a complication feared by surgeons, especially after extended hepatectomy or small liver transplant, although liver transplantation and liver resection are the first curative treatment for primary and secondary liver tumors [65][66]. PHLF and SFSS are characterized by hyperbilirubinemia, coagulopathy and ascites reflecting portal hyperperfusion, which occur within the first postoperative week. They can lead to post-operative sepsis and bleeding, increasing mortality and morbidity [67][68]. For living donor liver transplantation, the optimal future liver remnant or graft size depends on the graft-to-recipient weight ratio which must be above 0.8% [69]. In the context of extended hepatectomy, the optimal future liver remnant is mainly based on its volume, which must be >20% of the initial liver volume. Yet, conditions, such as steatosis, steatohepatitis, fibrosis, cirrhosis, chemotherapy-induced liver injury or cholestasis may overestimate the function of the liver remnant if only size is considered. If conditions mentioned above are not met, the risk for post-operative failure increases [69]. In such situations, another important role of LSEC becomes more apparent.34.1. What Causes Mortality after Extended Hepatectomy?
For years, it was supposed that the inability of the hepatocytes to proliferate after a SFSS-setting hepatectomy led to organ’s functional insufficiency. As a support, several teams described blunted or delayed hepatocyte proliferation after extended hepatectomy. Clavien’s group reported a decreased number of mitosis despite magnified Ki67 expression in extended versus 70% hepatectomy. High expression of p21, a cyclin-dependent kinase inhibitor, in the small remnant was found to be the cause of decreased hepatocyte proliferation as in p21 KO animals, hepatocyte proliferation was preserved, improving the animals’ survival[70]
. Moreover, failure to upregulate transcription factors (such as c-fos) necessary to drive cell cycle beyond G1 phase and delay of the proliferation phase has been observed in rats by other groups[71][72]. Failure of hepatocyte proliferation as the mechanism for SFSS and PHLF is opposed by several authors: Hepatocyte doublings after small for size setting hepatectomy in rats was ampler than after a well tolerated 70% liver resection
. Failure of hepatocyte proliferation as the mechanism for SFSS and PHLF is opposed by several authors: Hepatocyte doublings after small for size setting hepatectomy in rats was ampler than after a well tolerated 70% liver resection[73]
. Immunohistological studies confirmed high index of hepatocyte proliferation in small for size human grafts . These works suggest that PHLF and SFSS are not due to a failure of hepatocytes to proliferate (Figure 3).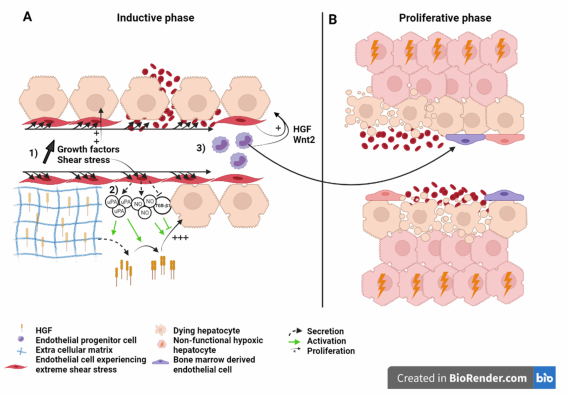
Figure 3. Liver sinusoid and endothelial cells during the (A) inductive phase and (B) angiogenic phase of liver regeneration
after extended hepatectomy.Scheme on the role of LSEC during regeneration after extended hepatectomy during the
inductive phase (A) where (1) growth factors, (2) severe shear stress, as well as (3) bone marrow endothelial progenitors
induce the proliferation of the hepatocytes. Severe shear stress induces sinusoidal denudation and hemorrhage. During
the proliferative phase (B), non-functional hypoxic hepatocytes and hemorrhage-induced necrosis lead to organ function
insufficiency and PHLF. Recruited endothelial progenitor cells become LSEC during regeneration. HGF: Hepatic growth
factor; NO: Nitric oxide; TGB-β1: Tumor growth factor beta 1; Wnt2: Wingless-type MMTV integration site family, member
2; HIF: Hypoxia inducible factor; VEGF: Vascular endothelial growth factor.
Liver sinusoid and endothelial cells during the (A) inductive phase and (B) angiogenic phase of liver regeneration after extended hepatectomy.Scheme on the role of LSEC during regeneration after extended hepatectomy during the inductive phase (A) where (1) growth factors, (2) severe shear stress, as well as (3) bone marrow endothelial progenitors induce the proliferation of the hepatocytes. Severe shear stress induces sinusoidal denudation and hemorrhage. During the proliferative phase (B), non-functional hypoxic hepatocytes and hemorrhage-induced necrosis lead to organ function insufficiency and PHLF. Recruited endothelial progenitor cells become LSEC during regeneration. HGF: Hepatic growth factor; NO: Nitric oxide; TGB-β1: Tumor growth factor beta 1; Wnt2: Wingless-type MMTV integration site family, member 2; HIF: Hypoxia inducible factor; VEGF: Vascular endothelial growth factor.
34.2. Liver Failure Because of Sinusoid Insufficiency?
The histological examination of the morphology of SFSS livers reveals the presence of endothelial denudation, hemorrhage, sinusoidal congestion and collapse of the space of Disse. Islets of hepatocytes (i.e., cluster of hepatocytes without interposition of sinusoids), and hepatocyte ballooning are also readily seen signing poor hepatocellular function[73][76].
Such observations support the fact that primary vascular damage is one of the causes of liver dysfunction. Hence, some authors suggested to rename the syndrome as “small for flow syndrome” with evidence that shear stress and perturbations of the microcirculation were significant contributors to the surgery-induced liver failure. Indeed, studies support that the
portal flow rather than the size of the liver remnant is the predictive factor for SFSS
[79]
. It was also associated with increased HGF concentrations and accelerated organ hypertrophy[68]
. Moreover, in the context of liver graft, providing that the portal pressure and flow are maintained under a given threshold, small livers regarded as too small for survival (defined by a graft-to-recipient weight ratio >0.8%) have been transplanted successfully[80]. The mechanism underlying functional failure and mortality in SFSS remains elusive. While the post-surgery increase in portal pressure and shear stress is needed to support regeneration (Figure 2), excessive portal pressure and excessive shear stress cause vascular damage and hepatocyte hyper-proliferation in extended resection that could be detrimental for the organ function. It has been suggested that islets of hepatocytes disconnected from the ordered sinusoidal organization experience a profound hypoxia leading to cell and organ dysfunction of the “SFSS liver remnant”
. The mechanism underlying functional failure and mortality in SFSS remains elusive. While the post-surgery increase in portal pressure and shear stress is needed to support regeneration (Figure 2), excessive portal pressure and excessive shear stress cause vascular damage and hepatocyte hyper-proliferation in extended resection that could be detrimental for the organ function. It has been suggested that islets of hepatocytes disconnected from the ordered sinusoidal organization experience a profound hypoxia leading to cell and organ dysfunction of the “SFSS liver remnant” . Hyper-proliferation of the hepatocytes, the fact that hepatocyte proliferation and sinusoidal cell proliferation are not in phase and the sinusoidal damage are three additive factors explaining that the growing mass of hepatocyte is improperly vascularized. Hepatocyte dysfunction and damage to the endothelium followed by hemorrhage in the liver parenchyma that can lead to necrosis, participate to liver failure[81] (Figure 3).
(Figure 3).34.3. Effect of the Modulation of Portal Hyperflow and Shear Stress after Extended Hepatectomy
The theory is supported by pre-clinical as well as clinical data showing that manipulations to reduce the portal flow, and thus, to mitigate the shear stress (hence endothelial damage and hepatocyte hyper-proliferation) in a SFSS-setting hepatectomy were effective in preventing liver failure. For instance, it has been reported that the ligation of the splenic artery (whether performed pre-, during, or immediately after surgery) reduced the portal flow by 52% and subsequently also mortality . Other techniques, such as splenectomy[84]
, splenorenal shunt[68]
, hemiporto-caval shunt[85] or mesocaval
shunt with ligation of the superior mesenteric artery
[86] to decrease portal flow successfully reduced mortality rates. Mechanical modulation of portal flow is currently being explored by Vibert’s team in a clinical trial (NCT02390713) where a pneumatic ring is used to modulate the diameter of the portal vein after major hepatectomy. This device precisely modulates the portal flow, as opposed to the techniques introduced immediately above. Pharmacological reduction of the portal flow has been reported to have a similar beneficial effect: Olprione, a phosphodiesterase inhibitor with vasodilating properties, demonstrated reduction in endothelial damage and hepatocyte apoptosis through the up-regulation of NO synthase in rats
to decrease portal flow successfully reduced mortality rates. Mechanical modulation of portal flow is currently being explored by Vibert’s team in a clinical trial (NCT02390713) where a pneumatic ring is used to modulate the diameter of the portal vein after major hepatectomy. This device precisely modulates the portal flow, as opposed to the techniques introduced immediately above. Pharmacological reduction of the portal flow has been reported to have a similar beneficial effect: Olprione, a phosphodiesterase inhibitor with vasodilating properties, demonstrated reduction in endothelial damage and hepatocyte apoptosis through the up-regulation of NO synthase in rats[87]
. Prostaglandin E1 also increased survival rates and liver regeneration[88]. Administration of NO donor FK 409 increased survival from 28.6% to 80%, an effect associated with decreased expression of Egr1, endothelin-1 and endothelin-1
receptor A and up–regulation of heme oxygenase-1
[89]
. Up- and down-regulation of these genes were also observed by Xu et al., using somatostatin in a rat model of orthotopic liver transplantation[90]
. More recently, Mokham et al., confirmed positive effects of the modulation of portal flow in pigs[91]
. It is anticipated that these procedures decrease the post-surgery hyperflow with, as a consequence, the preservation of the integrity of the sinusoids and the mitigation of the proliferative stimulus for hepatocytes. In support of this, slowing down hepatocyte regeneration with ERK1/2 et MEK inhibitor after 90% PHx in rats reduced the transient hepatocyte to LSEC numerical mismatch, maintained the liver architecture and improved the animal survival[73]. Therefore, the regulation of portal blood flow prevents post-operative failure by reducing hepatocyte proliferation (hence transiently avascular hepatocyte islets), as well as endothelial damage. Understanding whether the acceleration of angiogenesis and LSEC renewal as to match the high level of hepatocyte regeneration, and repair the sinusoidal damage would prevent SFSS, remains to be demonstrated. At the moment, there are no data available on the proliferation of LSEC or on cell types contributing to vascular remodeling after extended hepatectomy. In recent work, our team proposed experimental evidence that the stimulation of angiogenesis at early time points during regeneration of a small remnant prevented SFSS-induced mortality. Maneuvers, such as hepatic artery ligation concomitant to extended hepatectomy
or treatment with DMOG, a prolyl hydroxylase domain inhibitor that activates HIF-1α, triggered an early pro-angiogenic response and prevented the collapse of hepatic sinusoids in the small for size regenerating liver
[92]. Altogether, these pre-clinical and clinical data support the importance of remodeling the sinusoidal network according to hepatocyte proliferation during liver regeneration to maintain a functional lobular structure and sustain the metabolic activity of the proliferating hepatocytes. Mitigating the proliferative response after extended hepatectomy is beneficial to the patient’s life. In the same perspective, triggering early LSEC proliferation after extended hepatectomy may be useful in maintaining the organization of the lobule and the function of hepatocytes.
. Altogether, these pre-clinical and clinical data support the importance of remodeling the sinusoidal network according to hepatocyte proliferation during liver regeneration to maintain a functional lobular structure and sustain the metabolic activity of the proliferating hepatocytes. Mitigating the proliferative response after extended hepatectomy is beneficial to the patient’s life. In the same perspective, triggering early LSEC proliferation after extended hepatectomy may be useful in maintaining the organization of the lobule and the function of hepatocytes.45. Conclusions
Due to their location in the liver lobule, interposed between blood stream and hepatocytes, embraced by hepatic stellate cells and in physical contact with Kupffer cells, LSEC interact with and integrate an array of information from the environment. In this review, we presented research supporting the critical role of LSEC during liver regeneration. LSEC are necessary for the proliferation of hepatocyte and for the maintenance of an organized architecture of the lobule. Bone marrow- derived and native LSEC cooperate to play a role in the initiation, proliferative and termination phases of liver regeneration. The process becomes non-operational upon extended hepatectomy. Extreme and brutal increase in portal pressure leads to endothelial denudation with subsequent tissue necrosis and disturbance of the lobule structure. Regenerating hepatocytes do not have an organized vascular network along with which to align. Therefore, their function is compromised and leads to organ failure. The essential role of LSEC in liver regeneration designate them as attractive targets in reducing mortality. Surgical procedures and pharmaceutical treatments that decrease portal pressure also maintain the conventional lobular architecture with great results with respect to survival, both in animal and clinical studies. The need for a competent sinusoidal network to ensure proper function during regeneration supports the major role of LSEC and encourages more research targeting LSEC in liver regeneration.
References
- Stanger, B.Z. Cellular Homeostasis and Repair in the Mammalian Liver. Annu. Rev. Physiol. 2015, 77, 179–200.
- Wei, Y.; Wang, Y.G.; Jia, Y.; Li, L.; Yoon, J.; Zhang, S.; Wang, Z.; Zhang, Y.; Zhu, M.; Sharma, T.; et al. Liver homeostasis is maintained by midlobular zone 2 hepatocytes. Science 2021, 371, eabb1625.
- Michalopoulos, G.K. Liver regeneration. J. Cell. Physiol. 2007, 213, 286–300.
- Preziosi, M.E.; Monga, S.P. Update on the Mechanisms of Liver Regeneration. Semin. Liver Dis. 2017,37, 141–151.Preziosi, M.E.; Monga, S.P. Update on the Mechanisms of Liver Regeneration. Semin. Liver Dis. 2017, 37, 141–151.
- Michalopoulos, G.K.; DeFrances, M.C.; Cressman, D.E.; Greenbaum, L.E.; DeAngelis, R.A.; Ciliberto, G.; Furth, E.E.; Poli, V.; Taub, R. Liver Regeneration. Science 1997, 276, 60–66.
- Fausto, N. Liver regeneration. J. Hepatol. 2000, 32, 19–31.
- Fausto, N.; Campbell, J.S.; Riehle, K.J. Liver regeneration. Hepatology 2006, 43, S45–S53.
- Poisson, J.; Lemoinne, S.; Boulanger, C.M.; Durand, F.; Moreau, R.; Valla, D.; Rautou, P.-E. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J. Hepatol. 2017, 66, 212–227.
- Wang, L.; Wang, X.; Xie, G.; Wang, L.; Hill, C.K.; Deleve, L.D. Liver sinusoidal endothelial cell progenitor cells promote liver regeneration in rats. J. Clin. Investig. 2012, 122, 1567–1573.
- Deleve, L.D. Liver sinusoidal endothelial cells and liver regeneration. J. Clin. Investig. 2013, 123, 1861–1866.
- Sakamoto, T.; Liu, Z.; Murase, N.; Ezure, T.; Yokomuro, S.; Poli, V.; Demetris, A.J. Mitosis and apoptosis in the liver of interleukin-6-deficient mice after partial hepatectomy. Hepatology 1999, 29, 403–411.
- Sato, Y.; Koyama, S.; Tsukada, K.; Hatakeyama, K. Acute portal hypertension reflecting shear stress as a trigger of liver regeneration following partial hepatectomy. Surg. Today 1997, 27, 518–526.
- Schoen, J.M.; Wangb, H.H.; Minukac, G.Y.; Lautt, W. Shear Stress-Induced Nitric Oxide Release Triggers the Liver Regeneration Cascade. Nitric Oxide 2001, 5, 453–464.
- Albinsson, S.; Hellstrand, P. Integration of signal pathways for stretch-dependent growth and differentiation in vascular smooth muscle. Am. J. Physiol. Cell Physiol. 2007, 293, C772–C782.
- Weinbaum, S.; Zhang, X.; Han, Y.; Vink, H.; Cowin, S.C. Mechanotransduction and flow across the endothelial glycocalyx. Proc. Natl. Acad. Sci. USA 2003, 100, 7988–7995.
- Gracia-Sancho, J.; Russo, L.; García-Calderó, H.; Garcia-Pagan, J.C.; García-Cardeña, G.; Bosch, J. Endothelial expression of transcription factor Kruppel-like factor 2 and its vasoprotective target genes in the normal and cirrhotic rat liver. Gut 2010, 60, 517–524.
- Díaz-Juárez, J.A.; Hernández-Muñoz, R. Rat Liver Enzyme Release Depends on Blood Flow-Bearing Physical Forces Acting in Endothelium Glycocalyx rather than on Liver Damage. Oxidative Med. Cell. Longev. 2017, 2017, 1360565.
- Schoen, J.M.; Lautt, W.W. Nitric Oxide Potentiates C-Fos mRNA Expression after 2/3 Partial Hepatectomy. Proc. West. Pharmacol. Soc. 2002, 45, 47–48.
- Isomura, H.; Sawada, N.; Nakajima, Y.; Sakamoto, H.; Ikeda, T.; Kojima, T.; Enomoto, K.; Mori, M. Increase in portal flow induces c-myc expression in isolated perfused rat liver. J. Cell. Physiol. 1993, 154, 329–332.
- Gan, L.-M.; Doroudi, R.; Hägg, U.; Johansson, A.-M.; Selin-Sjögren, L.; Jern, S. Differential immediate-early gene responses to shear stress and intraluminal pressure in intact human conduit vessels. FEBS Lett. 2000, 477, 89–94.
- Lauber, D.T.; Tihanyi, D.K.; Czigány, Z.; Kovács, T.; Budai, A.; Drozgyik, D.; Fülöp, A.; Szijártó, A. Liver regeneration after different degrees of portal vein ligation. J. Surg. Res. 2016, 203, 451–458.
- Meier, M.; Andersen, K.J.; Knudsen, A.R.; Nyengaard, J.R.; Hamilton-Dutoit, S.; Mortensen, F.V. Liver regeneration is dependent on the extent of hepatectomy. J. Surg. Res. 2016, 205, 76–84.
- Shimazu, M.; Kato, Y.; Kawachi, S.; Tanabe, M.; Hoshino, K.; Wakabayashi, G.; Kitagawa, Y.; Kitajima, M. Impact of Portal Hemodynamic Changes in Partial Liver Grafts on Short-Term Graft Regeneration in Living Donor Liver Transplantation. Transplant. Proc. 2016, 48, 2747–2755.
- Eguchi, S.; Yanaga, K.; Sugiyama, N.; Okudaira, S.; Furui, J.; Kanematsu, T. Relationship between portal venous flow and liver regeneration in patients after living donor right-lobe liver transplantation. Liver Transplant. 2003, 9, 547–551.
- Oyama, T.; Sadamori, H.; Matsukawa, H.; Murata, H.; Umeda, Y.; Watanabe, Y.; Ozaki, M.; Iwagaki, H.; Tanaka, N.; Yagi, T. Small liver graft regenerates through immediate increase of HGF and IL-6--possible involvement of sinusoidal tensile/shear stress in small liver graft. Hepatogastroenterology 2008, 54, 2078–2083.
- Jones, D.E.; Tran-Patterson, R.; Cui, D.M.; Davin, D.; Estell, K.P.; Miller, D.M. Epidermal growth factor secreted from the salivary gland is necessary for liver regeneration. Am. J. Physiol. Gastrointest. Liver Physiol. 1995, 268, G872–G878.
- Rubin, R.A.; O’Keefe, E.J.; Earp, H.S. Alteration of epidermal growth factor-dependent phosphorylation during rat liver regeneration. Proc. Natl. Acad. Sci. USA 1982, 79, 776–780.
- Olsen, P.S.; Boesby, S.; Kirkegaard, P.; Therkelsen, K.; Almdal, T.; Poulsen, S.S.; Nexø, E. Influence of epidermal growth factor on liver regeneration after partial hepatectomy in rats. Hepatology 1988, 8, 992–996.
- Shavandi, A.; Saeedi, P.; Gérard, P.; Jalalvandi, E.; Cannella, D.; Bekhit, A.E. The role of microbiota in tissue repair and regeneration. J. Tissue Eng. Regen. Med. 2020, 14, 539–555.
- Shah, V.; Haddad, F.G.; Garcia-Cardena, G.; Frangos, J.A.; Mennone, A.; Groszmann, R.J.; Sessa, W.C. Liver sinusoidal endothelial cells are responsible for nitric oxide modulation of resistance in the hepatic sinusoids. J. Clin. Investig. 1997, 100, 2923–2930.
- Patijn, G.A.; Lieber, A.; Schowalter, D.B.; Schwall, R.; Kay, M.A. Hepatocyte growth factor induces hepatocyte proliferationin vivo and allows for efficient retroviral-mediated gene transfer in mice. Hepatology 1998, 28, 707–716.
- Sokabe, T.; Yamamoto, K.; Ohura, N.; Nakatsuka, H.; Qin, K.; Obi, S.; Kamiya, A.; Ando, J. Differential regulation of urokinase-type plasminogen activator expression by fluid shear stress in human coronary artery endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2027–H2034.
- Dolan, J.M.; Sim, F.J.; Meng, H.; Kolega, J. Endothelial cells express a unique transcriptional profile under very high wall shear stress known to induce expansive arterial remodeling. Am. J. Physiol. Cell Physiol. 2012, 302, C1109–C1118.
- Kim, T.; Mars, W.M.; Stolz, D.B.; Petersen, B.E.; Michalopoulos, G.K. Extracellular matrix remodeling at the early stages of liver regeneration in the rat. Hepatology 1997, 26, 896–904.
- Nejak-Bowen, K.; Orr, A.; Bowen, W.C., Jr.; Michalopoulos, G.K. Conditional Genetic Elimination of Hepatocyte Growth Factor in Mice Compromises Liver Regeneration after Partial Hepatectomy. PLoS ONE 2013, 8, e59836.
- Mars, W.M.; Zarnegar, R.; Michalopoulos, G.K. Activation of Hepatocyte Growth Factor by the Plasminogen Activators uPA and tPA. Am. J. Pathol. 1993, 143, 949–958.
- Mars, W.M.; Kim, T.H.; Stolz, D.B.; Liu, M.L.; Michalopoulos, G.K. Presence of urokinase in serum-free primary rat hepatocyte cultures and its role in activating hepatocyte growth factor. Cancer Res. 1996, 56, 2837–2843.
- Shanmukhappa, K.; Sabla, G.E.; Degen, J.L.; Bezerra, J.A. Urokinase-type plasminogen activator supports liver repair independent of its cellular receptor. BMC Gastroenterol. 2006, 6, 40.
- Ding, B.-S.; Nolan, D.J.; Butler, J.M.; James, D.; Babazadeh, A.O.; Rosenwaks, Z.; Mittal, V.; Kobayashi, H.; Shido, K.; Lyden, D.; et al. Inductive angiocrine signals from sinusoidal endothelium are required for liver regeneration. Nat. Cell Biol. 2010, 468, 310–315.
- Döme, B.; Dobos, J.; Tóvári, J.; Paku, S.; Kovács, G.; Ostoros, G.; Tímár, J. Circulating bone marrow-derived endothelial progenitor cells: Characterization, mobilization, and therapeutic considerations in malignant disease. Cytom. Part A 2008, 73, 186–193.
- Timmermans, F.; Plum, J.; Yoder, M.; Ingram, D.A.; Vandekerckhove, B.; Case, J. Endothelial progenitor cells: Identity defined? J. Cell. Mol. Med. 2008, 13, 87–102.
- Hristov, M.; Erl, W.; Weber, P.C. Endothelial progenitor cells: Mobilization, differentiation, and homing. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1185–1189.
- Fujii, H.; Hirose, T.; Oe, S.; Yasuchika, K.; Azuma, H.; Fujikawa, T.; Nagao, M.; Yamaoka, Y. Contribution of bone marrow cells to liver regeneration after partial hepatectomy in mice. J. Hepatol. 2002, 36, 653–659.
- Li, N.; Hua, J. Immune cells in liver regeneration. Oncotarget 2016, 8, 3628–3639.
- Harb, R.; Xie, G.; Lutzko, C.; Guo, Y.; Wang, X.; Hill, C.K.; Kanel, G.C.; DeLeve, L.D. Bone Marrow Progenitor Cells Repair Rat Hepatic Sinusoidal Endothelial Cells After Liver Injury. Gastroenterology 2009, 137, 704–712.
- Deleve, L.D.; Wang, X.; Wang, L. VEGF-sdf1 recruitment of CXCR7 bone marrow progenitors of liver sinusoidal endothelial cells promotes rat liver regeneration. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, 739–746.
- Odent (Grigorescu), G.; Rosca, A.-M.; Preda, M.B.; Tutuianu, R.; Simionescu, M.; Burlacu, A. Synergic effects of VEGF-A and SDF-1 on the angiogenic properties of endothelial progenitor cells. J. Tissue Eng. Regen. Med. 2016, 11, 3241–3252.
- Moore, M.A.; Hattori, K.; Heissig, B.; Shieh, J.-H.; Dias, S.; Crystal, R.G.; Rafii, S. Mobilization of Endothelial and Hematopoietic Stem and Progenitor Cells by Adenovector-Mediated Elevation of Serum Levels of SDF-1, VEGF, and Angiopoietin-1. Ann. N. Y. Acad. Sci. 2006, 938, 36–47.
- Peled, A.; Grabovsky, V.; Habler, L.; Sandbank, J.; Arenzana-Seisdedos, F.; Petit, I.; Ben-Hur, H.; Lapidot, T.; Alon, R. The chemokine SDF-1 stimulates integrin-mediated arrest of CD34+ cells on vascular endothelium under shear flow. J. Clin. Investig. 1999, 104, 1199–1211.
- Silverman, M.D.; Haas, C.S.; Rad, A.M.; Arbab, A.S.; Koch, A.E. The role of vascular cell adhesion molecule 1/very late activation antigen 4 in endothelial progenitor cell recruitment to rheumatoid arthritis synovium. Arthritis Rheum. 2007, 56, 1817–1826.
- Yoon, C.-H.; Hur, J.; Oh, I.-Y.; Park, K.-W.; Kim, T.-Y.; Shin, J.-H.; Kim, J.-H.; Lee, C.-S.; Chung, J.-K.; Park, Y.-B.; et al. Intercellular Adhesion Molecule-1 is Upregulated in Ischemic Muscle, Which Mediates Trafficking of Endothelial Progenitor Cells. Arter. Thromb. Vasc. Biol. 2006, 26, 1066–1072.
- Jin, H.; Aiyer, A.; Su, J.; Borgström, P.; Stupack, D.; Friedlander, M.; Varner, J. A homing mechanism for bone marrow–derived progenitor cell recruitment to the neovasculature. J. Clin. Investig. 2006, 116, 652–662.
- Chavakis, E.; Aicher, A.; Heeschen, C.; Sasaki, K.-I.; Kaiser, R.; El Makhfi, N.; Urbich, C.; Peters, T.; Scharffetter-Kochanek, K.; Zeiher, A.M.; et al. Role of β2-integrins for homing and neovascularization capacity of endothelial progenitor cells. J. Exp. Med. 2004, 201, 63–72.
- Tilling, L.; Chowienczyk, P.; Clapp, B. Progenitors in motion: Mechanisms of mobilization of endothelial progenitor cells. Br. J. Clin. Pharmacol. 2009, 68, 484–492. Tilling, L.; Chowienczyk, P.; Clapp, B. Progenitors in motion: Mechanisms of mobilization of endothelial progenitor cells. Br. J. Clin. Pharmacol. 2009, 68, 484–492.
- Myronovych, A.; Murata, S.; Chiba, M.; Matsuo, R.; Ikeda, O.; Watanabe, M.; Hisakura, K.; Nakano, Y.; Kohno, K.; Kawasaki, T.; et al. Role of platelets on liver regeneration after 90% hepatectomy in mice. J. Hepatol. 2008, 49, 363–372.
- Meyer, J.; Lejmi, E.; Fontana, P.; Morel, P.; Gonelle-Gispert, C.; Bühler, L. A focus on the role of platelets in liver regeneration: Do platelet-endothelial cell interactions initiate the regenerative process? J. Hepatol. 2015, 63, 1263–1271.
- Melgar-Lesmes, P.; Edelman, E. Monocyte-endothelial cell interactions in the regulation of vascular sprouting and liver regeneration in mouse. J. Hepatol. 2015, 63, 917–925.
- Hu, J.; Srivastava, K.; Wieland, M.; Runge, A.; Mogler, C.; Besemfelder, E.; Terhardt, D.; Vogel, M.J.; Cao, L.; Korn, C.; et al. Endothelial Cell-Derived Angiopoietin-2 Controls Liver Regeneration as a Spatiotemporal Rheostat. Science 2014, 343, 416–419.
- Taniguchi, E.; Sakisaka, S.; Matsuo, K.; Tanikawa, K.; Sata, M. Expression and role of vascular endothelial growth factor in liver regeneration after partial hepatectomy in rats. J. Histochem. Cytochem. 2001, 49, 121–129.
- Shimizu, H.; Miyazaki, M.; Wakabayashi, Y.; Mitsuhashi, N.; Kato, A.; Ito, H.; Nakagawa, K.; Yoshidome, H.; Kataoka, M.; Nakajima, N. Vascular endothelial growth factor secreted by replicating hepatocytes induces sinusoidal endothelial cell proliferation during regeneration after partial hepatectomy in rats. J. Hepatol. 2001, 34, 683–689.
- Furnus, C.; Inda, A.; Andrini, L.; García, M.; García, A.; Badrán, A.; Errecalde, A. Chronobiology of the proliferative events related to angiogenesis in mice liver regeneration after partial hepatectomy. Cell Biol. Int. 2003, 27, 383–386.
- Kron, P.; Linecker, M.; Limani, P.; Schlegel, A.; Kambakamba, P.; Lehn, J.-M.; Nicolau, C.; Graf, R.; Humar, B.; Clavien, P.-A. Hypoxia-driven Hif2α coordinates mouse liver regeneration by coupling parenchymal growth to vascular expansion. Hepatology 2016, 64, 2198–2209.
- Braun, L.; Mead, J.E.; Panzica, M.; Mikumo, R.; Bell, G.I.; Fausto, N. Transforming growth factor beta mRNA increases during liver regeneration: A possible paracrine mechanism of growth regulation. Proc. Natl. Acad. Sci. USA 1988, 85, 1539–1543.
- Michalopoulos, G.K.; Bhushan, B. Liver regeneration: Biological and pathological mechanisms and implications. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 40–55.
- Poon, R.T.; Fan, S.T.; Lo, C.M.; Liu, C.L.; Lam, C.M.; Yuen, W.K.; Yeung, C.; Wong, J.; Nagorney, D.M.; Henderson, J.M.; et al. Improving perioperative outcome expands the role of hepatectomy in management of benign and malignant hepatobiliary diseases: Analysis of 1222 consecutive patients from a prospective database. Ann. Surg. 2004, 240, 698–710.
- Imamura, H.; Seyama, Y.; Kokudo, N.; Aoki, T.; Sano, K.; Minagawa, M.; Sugawara, Y.; Makuuchi, M. Single and multiple resections of multiple hepatic metastases of colorectal origin. Surgery 2004, 135, 508–517.
- Dahm, F.; Georgiev, P.; Clavien, P.-A. Small-for-Size Syndrome after Partial Liver Transplantation: Definition, Mechanisms of Disease and Clinical Implications. Arab. Archaeol. Epigr. 2005, 5, 2605–2610. Dahm, F.; Georgiev, P.; Clavien, P.-A. Small-for-Size Syndrome after Partial Liver Transplantation: Definition, Mechanisms of Disease and Clinical Implications. Arab. Archaeol. Epigr. 2005, 5, 2605–2610.
- Yagi, S.; Iida, T.; Hori, T.; Taniguchi, K.; Yamamoto, C.; Yamagiwa, K.; Uemoto, S. Optimal Portal Venous Circulation for Liver Graft Function after Living-Donor Liver Transplantation. Transplantation 2006, 81, 373–378.
- Guglielmi, A.; Ruzzenente, A.; Conci, S.; Valdegamberi, A.; Iacono, C. How Much Remnant Is Enough in Liver Resection? Dig. Surg. 2012, 29, 6–17.
- Lehmann, K.; Tschuor, C.; Rickenbacher, A.; Jang, J.; Oberkofler, C.E.; Tschopp, O.; Schultze, S.M.; Raptis, D.A.; Weber, A.; Graf, R.; et al. Liver Failure After Extended Hepatectomy in Mice Is Mediated by a p21-Dependent Barrier to Liver Regeneration. Gastroenterology 2012, 143, 1609–1619.e4.
- Zieve, L.; Anderson, W.R.; Lindblad, S. Course of hepatic regeneration after 80% to 90% resection of normal rat liver. Comparison with two-lobe and one-lobe hepatectomy. J. Lab. Clin. Med. 1985, 105, 331–336.
- Moser, M.J.; Gong, Y.; Zhang, M.N.; Johnston, J.; Lipschitz, J.; Minuk, G.Y. Immediate-Early Protooncogene Expression and Liver Function Following Various Extents of Partial Hepatectomy in the Rat. Dig. Dis. Sci. 2001, 46, 907–914.
- Ninomiya, M.; Shirabe, K.; Terashi, T.; Ijichi, H.; Yonemura, Y.; Harada, N.; Soejima, Y.; Taketomi, A.; Shimada, M.; Maehara, Y. Deceleration of Regenerative Response Improves the Outcome of Rat with Massive Hepatectomy. Arab. Archaeol. Epigr. 2010, 10, 1580–1587. Ninomiya, M.; Shirabe, K.; Terashi, T.; Ijichi, H.; Yonemura, Y.; Harada, N.; Soejima, Y.; Taketomi, A.; Shimada, M.; Maehara, Y. Deceleration of Regenerative Response Improves the Outcome of Rat with Massive Hepatectomy. Arab. Archaeol. Epigr. 2010, 10, 1580–1587.
- Gridelli, B.G.; Gruttadauria, S.; Pagano, D.; Liotta, R.; Tropea, A.; Tuzzolino, F.; Marrone, G.; Mamone, G.; Marsh, J.W.; Miraglia, R.; et al. Liver Volume Restoration and Hepatic Microarchitecture in Small-for-Size Syndrome. Ann. Transplant. 2015, 20, 381–389.
- Byun, S.H.; Yang, H.S.; Kim, J.H. Liver graft hyperperfusion in the early postoperative period promotes hepatic regeneration 2 weeks after living donor liver transplantation. Medicine 2016, 95, e5404.
- Demetris, A.J.; Kelly, D.M.; Eghtesad, B.; Fontes, P.; Marsh, J.W.; Tom, K.; Tan, H.P.; Shaw-Stiffel, T.; Boig, L.; Novelli, P.; et al. Pathophysiologic Observations and Histopathologic Recognition of the Portal Hyperperfusion or Small-for-Size Syndrome. Am. J. Surg. Pathol. 2006, 30, 986–993.
- Vasavada, B.; Chen, C.L.; Zakaria, M. Portal flow is the main predictor of early graft dysfunction regardless of the GRWR status in living donor liver transplantation—A retrospective analysis of 134 patients. Int. J. Surg. 2014, 12, 177–180.
- Asencio, J.; Vaquero, J.; Olmedilla, L.; Sabrido, J.G. “Small-for-flow” syndrome: Shifting the “size” paradigm. Med. Hypotheses 2013, 80, 573–577.
- Allard, M.-A.; Adam, R.; Bucur, P.-O.; Termos, S.; Cunha, A.S.; Bismuth, H.; Castaing, D.; Vibert, E. Posthepatectomy Portal Vein Pressure Predicts Liver Failure and Mortality after Major Liver Resection on Noncirrhotic Liver. Ann. Surg. 2013, 258, 822–830.
- Brown, R.S. Live Donors in Liver Transplantation. Gastroenterology 2008, 134, 1802–1813.
- Golse, N.; Bucur, P.O.; Adam, R.; Castaing, D.; Cunha, A.S.; Vibert, E. New Paradigms in Post-hepatectomy Liver Failure. J. Gastrointest. Surg. 2012, 17, 593–605.
- Umeda, Y.; Yagi, T.; Sadamori, H.; Fujiwara, T. Small-for-Size Syndrome after Living Donor Liver Transplantation. In Liver Transplantation: Technical Issues and Complications; Abdeldayem, H., Ed.; IntechOpen: London, UK, 2012; ISBN 978-953-51-0015-7.
- Troisi, R.; Ricciardi, S.; Smeets, P.; Petrovic, M.; Van Maele, G.; Colle, I.; Van Vlierberghe, H.; De Hemptinne, B. Effects of Hemi-Portocaval Shunts for Inflow Modulation on the Outcome of Small-for-Size Grafts in Living Donor Liver Transplantation. Arab. Archaeol. Epigr. 2005, 5, 1397–1404.
- Sato, Y.; Yamamoto, S.; Oya, H.; Nakatsuka, H.; Tsukahara, A.; Kobayashi, T.; Watanabe, T.; Hatakeyama, K. Sple-nectomy for reduction of excessive portal hypertension after adult living-related donor liver transplantation. Hepatogastroenterology 2002, 49, 1652–1655.
- Yamada, T.; Tanaka, K.; Uryuhara, K.; Ito, K.; Takada, Y.; Uemoto, S. Selective Hemi-Portocaval Shunt Based on Portal Vein Pressure for Small-for-Size Graft in Adult Living Donor Liver Transplantation. Arab. Archaeol. Epigr. 2008, 8, 847–853.
- Boillot, O. Portomesenteric disconnection for small-for-size grafts in liver transplantation: Preclinical studies in pigs. Liver Transplant. 2003, 9, S42–S46.
- Yamanaka, K.; Hatano, E.; Narita, M.; Kitamura, K.; Yanagida, A.; Asechi, H.; Nagata, H.; Taura, K.; Nitta, T.; Uemoto, S. Olprinone attenuates excessive shear stress through up-regulation of endothelial nitric oxide synthase in a rat excessive hepatectomy model. Liver Transplant. 2010, 17, 60–69.
- Morioka, D.; Matsuo, K.; Endo, I.; Togo, S.; Kubota, T.; Sekido, H.; Saito, S.; Ichikawa, Y.; Shimada, H. Prostaglandin E1 improved the function of transplanted fatty liver in a rat reduced-size-liver transplantation model under conditions of permissible cold preservation. Liver Transplant. 2003, 9, 79–86.
- Man, K.; Lee, K.W.; Liang, T.B.; Lo, C.M.; Fung, P.C.-W.; Tsui, S.H.; Li, X.L.; Ng, K.T.-P.; Fan, S.T. FK 409 Ameliorates Small-for-Size Liver Graft Injury by Attenuation of Portal Hypertension and Down-Regulation of Egr-1 Pathway. Ann. Surg. 2004, 240, 159–168.
- Xu, X.; Man, K.; Zheng, S.S.; Liang, T.B.; Lee, K.W.; Ng, K.T.-P.; Fan, S.T.; Lo, C.M. Attenuation of acute phase shear stress by somatostatin improves small-for-size liver graft survival. Liver Transplant. 2006, 12, 621–627.
- Mohkam, K.; Darnis, B.; Schmitt, Z.; Duperret, S.; Ducerf, C.; Mabrut, J.-Y. Successful modulation of portal inflow by somatostatin in a porcine model of small-for-size syndrome. Am. J. Surg. 2016, 212, 321–326.
- Dili, A.; Bertrand, C.; Lebrun, V.; Pirlot, B.; Leclercq, I.A. Hypoxia protects the liver from Small for Size Syndrome: A lesson learned from the associated liver partition and portal vein ligation for staged hepatectomy (ALPPS) procedure in rats. Arab. Archaeol. Epigr. 2019, 19, 2979–2990.
- Braun, L.; Mead, J.E.; Panzica, M.; Mikumo, R.; Bell, G.I.; Fausto, N. Transforming growth factor beta mRNA increases during liver regeneration: A possible paracrine mechanism of growth regulation. Proc. Natl. Acad. Sci. USA 1988, 85, 1539–1543.
- Michalopoulos, G.K.; Bhushan, B. Liver regeneration: Biological and pathological mechanisms and implications. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 40–55.
- Poon, R.T.; Fan, S.T.; Lo, C.M.; Liu, C.L.; Lam, C.M.; Yuen, W.K.; Yeung, C.; Wong, J.; Nagorney, D.M.; Henderson, J.M.; et al. Improving perioperative outcome expands the role of hepatectomy in management of benign and malignant hepatobiliary diseases: Analysis of 1222 consecutive patients from a prospective database. Ann. Surg. 2004, 240, 698–710.
- Imamura, H.; Seyama, Y.; Kokudo, N.; Aoki, T.; Sano, K.; Minagawa, M.; Sugawara, Y.; Makuuchi, M. Single and multiple resections of multiple hepatic metastases of colorectal origin. Surgery 2004, 135, 508–517.
- Dahm, F.; Georgiev, P.; Clavien, P.-A. Small-for-Size Syndrome after Partial Liver Transplantation: Definition, Mechanisms of Disease and Clinical Implications. Arab. Archaeol. Epigr. 2005, 5, 2605–2610.
- Yagi, S.; Iida, T.; Hori, T.; Taniguchi, K.; Yamamoto, C.; Yamagiwa, K.; Uemoto, S. Optimal Portal Venous Circulation for Liver Graft Function after Living-Donor Liver Transplantation. Transplantation 2006, 81, 373–378.
- Guglielmi, A.; Ruzzenente, A.; Conci, S.; Valdegamberi, A.; Iacono, C. How Much Remnant Is Enough in Liver Resection? Dig. Surg. 2012, 29, 6–17.
- Lehmann, K.; Tschuor, C.; Rickenbacher, A.; Jang, J.; Oberkofler, C.E.; Tschopp, O.; Schultze, S.M.; Raptis, D.A.; Weber, A.; Graf, R.; et al. Liver Failure After Extended Hepatectomy in Mice Is Mediated by a p21-Dependent Barrier to Liver Regeneration. Gastroenterology 2012, 143, 1609–1619.e4.
- Zieve, L.; Anderson, W.R.; Lindblad, S. Course of hepatic regeneration after 80% to 90% resection of normal rat liver. Comparison with two-lobe and one-lobe hepatectomy. J. Lab. Clin. Med. 1985, 105, 331–336.
- Moser, M.J.; Gong, Y.; Zhang, M.N.; Johnston, J.; Lipschitz, J.; Minuk, G.Y. Immediate-Early Protooncogene Expression and Liver Function Following Various Extents of Partial Hepatectomy in the Rat. Dig. Dis. Sci. 2001, 46, 907–914.
- Ninomiya, M.; Shirabe, K.; Terashi, T.; Ijichi, H.; Yonemura, Y.; Harada, N.; Soejima, Y.; Taketomi, A.; Shimada, M.; Maehara, Y. Deceleration of Regenerative Response Improves the Outcome of Rat with Massive Hepatectomy. Arab. Archaeol. Epigr. 2010, 10, 1580–1587.
- Gridelli, B.G.; Gruttadauria, S.; Pagano, D.; Liotta, R.; Tropea, A.; Tuzzolino, F.; Marrone, G.; Mamone, G.; Marsh, J.W.; Miraglia, R.; et al. Liver Volume Restoration and Hepatic Microarchitecture in Small-for-Size Syndrome. Ann. Transplant. 2015, 20, 381–389.
- Byun, S.H.; Yang, H.S.; Kim, J.H. Liver graft hyperperfusion in the early postoperative period promotes hepatic regeneration 2 weeks after living donor liver transplantation. Medicine 2016, 95, e5404.
- Demetris, A.J.; Kelly, D.M.; Eghtesad, B.; Fontes, P.; Marsh, J.W.; Tom, K.; Tan, H.P.; Shaw-Stiffel, T.; Boig, L.; Novelli, P.; et al. Pathophysiologic Observations and Histopathologic Recognition of the Portal Hyperperfusion or Small-for-Size Syndrome. Am. J. Surg. Pathol. 2006, 30, 986–993.
- Vasavada, B.; Chen, C.L.; Zakaria, M. Portal flow is the main predictor of early graft dysfunction regardless of the GRWR status in living donor liver transplantation—A retrospective analysis of 134 patients. Int. J. Surg. 2014, 12, 177–180.
- Asencio, J.; Vaquero, J.; Olmedilla, L.; Sabrido, J.G. “Small-for-flow” syndrome: Shifting the “size” paradigm. Med. Hypotheses 2013, 80, 573–577.
- Allard, M.-A.; Adam, R.; Bucur, P.-O.; Termos, S.; Cunha, A.S.; Bismuth, H.; Castaing, D.; Vibert, E. Posthepatectomy Portal Vein Pressure Predicts Liver Failure and Mortality after Major Liver Resection on Noncirrhotic Liver. Ann. Surg. 2013, 258, 822–830.
- Brown, R.S. Live Donors in Liver Transplantation. Gastroenterology 2008, 134, 1802–1813.
- Golse, N.; Bucur, P.O.; Adam, R.; Castaing, D.; Cunha, A.S.; Vibert, E. New Paradigms in Post-hepatectomy Liver Failure. J. Gastrointest. Surg. 2012, 17, 593–605.
- Umeda, Y.; Yagi, T.; Sadamori, H.; Fujiwara, T. Small-for-Size Syndrome after Living Donor Liver Transplantation. In Liver Transplantation: Technical Issues and Complications; Abdeldayem, H., Ed.; IntechOpen: London, UK, 2012; ISBN 978-953-51-0015-7.
- Troisi, R.; Ricciardi, S.; Smeets, P.; Petrovic, M.; Van Maele, G.; Colle, I.; Van Vlierberghe, H.; De Hemptinne, B. Effects of Hemi-Portocaval Shunts for Inflow Modulation on the Outcome of Small-for-Size Grafts in Living Donor Liver Transplantation. Arab. Archaeol. Epigr. 2005, 5, 1397–1404.
- Sato, Y.; Yamamoto, S.; Oya, H.; Nakatsuka, H.; Tsukahara, A.; Kobayashi, T.; Watanabe, T.; Hatakeyama, K. Sple-nectomy for reduction of excessive portal hypertension after adult living-related donor liver transplantation. Hepatogastroenterology 2002, 49, 1652–1655.
- Yamada, T.; Tanaka, K.; Uryuhara, K.; Ito, K.; Takada, Y.; Uemoto, S. Selective Hemi-Portocaval Shunt Based on Portal Vein Pressure for Small-for-Size Graft in Adult Living Donor Liver Transplantation. Arab. Archaeol. Epigr. 2008, 8, 847–853.
- Boillot, O. Portomesenteric disconnection for small-for-size grafts in liver transplantation: Preclinical studies in pigs. Liver Transplant. 2003, 9, S42–S46.
- Yamanaka, K.; Hatano, E.; Narita, M.; Kitamura, K.; Yanagida, A.; Asechi, H.; Nagata, H.; Taura, K.; Nitta, T.; Uemoto, S. Olprinone attenuates excessive shear stress through up-regulation of endothelial nitric oxide synthase in a rat excessive hepatectomy model. Liver Transplant. 2010, 17, 60–69.
- Morioka, D.; Matsuo, K.; Endo, I.; Togo, S.; Kubota, T.; Sekido, H.; Saito, S.; Ichikawa, Y.; Shimada, H. Prostaglandin E1 improved the function of transplanted fatty liver in a rat reduced-size-liver transplantation model under conditions of permissible cold preservation. Liver Transplant. 2003, 9, 79–86.
- Man, K.; Lee, K.W.; Liang, T.B.; Lo, C.M.; Fung, P.C.-W.; Tsui, S.H.; Li, X.L.; Ng, K.T.-P.; Fan, S.T. FK 409 Ameliorates Small-for-Size Liver Graft Injury by Attenuation of Portal Hypertension and Down-Regulation of Egr-1 Pathway. Ann. Surg. 2004, 240, 159–168.
- Xu, X.; Man, K.; Zheng, S.S.; Liang, T.B.; Lee, K.W.; Ng, K.T.-P.; Fan, S.T.; Lo, C.M. Attenuation of acute phase shear stress by somatostatin improves small-for-size liver graft survival. Liver Transplant. 2006, 12, 621–627.
- Mohkam, K.; Darnis, B.; Schmitt, Z.; Duperret, S.; Ducerf, C.; Mabrut, J.-Y. Successful modulation of portal inflow by somatostatin in a porcine model of small-for-size syndrome. Am. J. Surg. 2016, 212, 321–326.
- Dili, A.; Bertrand, C.; Lebrun, V.; Pirlot, B.; Leclercq, I.A. Hypoxia protects the liver from Small for Size Syndrome: A lesson learned from the associated liver partition and portal vein ligation for staged hepatectomy (ALPPS) procedure in rats. Arab. Archaeol. Epigr. 2019, 19, 2979–2990.