Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Jose Antonio Curiel and Version 2 by Rita Xu.
Fucosylated carbohydrates and glycoproteins from human breast milk are essential for the development of the gut microbiota in early life because they are selectively metabolized by bifidobacteria.
- bifidobacteria
- fucosidases
- glycosyl hydrolases
1. Introduction
The impact of human milk glycobiome on the gut microbiota of infants is well established [1]. While a great part of the components of breast milk provide nutrients to the infant, human milk oligosaccharides (HMOs) and human milk glycoproteins (HMGs) selectively favor the colonization and growth of bifidobacteria in the infant intestine, contributing to the development of the gut microbiota [1][2][1,2]. In this regard, Bifidobacterium species are considered key actors in the multifaceted process of gut development and maturation of the immune system [3]. In fact, during the first months of birth, the loss of bifidobacteria or the gain of other bacteria can significantly alter the progression of the healthy microbial community with negative consequences for the infant, including a predisposition to autoimmune and/or metabolic diseases such as allergies and childhood obesity [4][5][4,5]. Concerning to that, fucosylated HMOs (FHMOs) and fucosylated HMGs (FHMGs) constitute a great part of the glycobiome of the breast milk [6] (Figure 1) and have been proposed to be essential in the development of the microbiota [7].
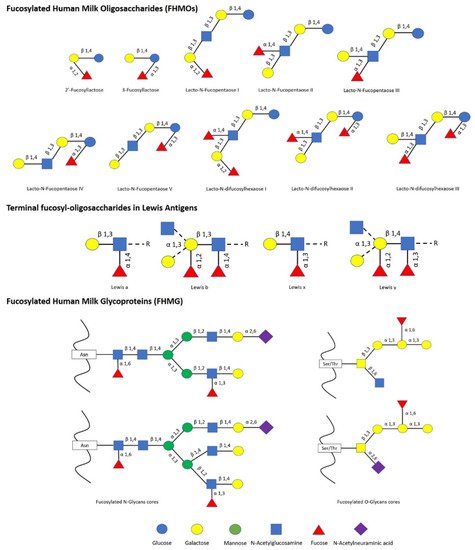
FHMOs constitute the largest fraction of human milk oligosaccharides, and although they show a small number of different conformations, they can make up to 70% of the total in an individual mother’s milk [6]. The fucosylated trisaccharide 2′-fucosyllactose is the most abundant FHMO, representing from 12 to 45% of the total HMO content in breastmilk, while 3-fucosyllactose is less abundant, from 0.5% to 3% [8]. On the other hand, there are several FMHGs investigated, and contrary to FHMOs, they appear at lower concentration but show a higher number of different forms, including lactoferrin (17%), immunoglobulins IgG (<1%), IgM (<1%), and secretory IgA (11%) [9][10][11][9,10,11]. Both FHMOs and FHMGs stand out for their ability to stimulate the growth of bifidobacteria [7][12][7,12], whose metabolism transforms fucosylated oligosaccharides into short-chain fatty acids (SCFAs) such as acetate, formate, lactate, and pyruvate [13], which in turn stimulate the immune system by inducing the differentiation of T-regulatory cells via inhibition of histone deacetylase [14].
The great influence of fucosylated compounds present in breast milk on bifidobacteria is due to their ability to metabolize them, being α-L-fucosidases (henceforth, fucosidases) indispensable tools that allow shaping the gut microbiome in the first months of life.
According to CAZy database, hitherto, more than 10,000 sequences have been identified in silico as α-L-fucosidases, belonging to a wide variety of organisms from archaea to fungi and plants. However, the vast majority of fucosidase sequences have been described in bacteria and belong to more than 2000 bacteria species (www.cazy.org). This database classifies fucosidases into three families (GH29, GH95, and GH151) according to their catalytic structures. GH29 fucosidases act through a retaining mechanism and have a broader substrate specificity, including hydrolysis of Fuc-α1,3/4/6 linkages [15]. Moreover, family GH29 fucosidases have been subclassified into two subfamilies. The subfamily A contains α-fucosidases with relatively relaxed substrate specificities, able to hydrolyze p-nitrophenyl-α-L-fucopyranoside (pNP-fucose), while the members of subfamily B are specific to α1,3/4-glycosidic linkages and are practically unable to hydrolyze pNP-fucose [16]. Although GH29 fucosidases also could exhibit hydrolysis of Fuc-α1,2 linkages, that activity is mainly attributed to GH95 family, which catalyzes the hydrolysis of fucose linkages through an inverting mechanism, resulting in the inversion of the anomeric configuration [17][18][17,18]. Finally, GH151 family has poor activity on fucosylated substrates; this is the reason why it is currently questioned as to whether they are genuine fucosidases [19][20][21][19,20,21].
Even though species of the Bifidobacterium genus dominate the infant gut microbiota in early life, and given the importance of their metabolism of fucosylated conjugates, there are only a few bifidobacterial species studied extensively at both cellular and genomics level for their ability to utilize fucosylated carbohydrates, including B. bifidum and Bifidobacterium longum subsp. infantis [22][23][22,23]. However, different strain-dependent metabolic abilities have been unraveled for the use of fucosylated conjugates and are likely determined by their fucosidases’ diversity [24]. Indeed, agreeing with the evolution and phylogenetics of fucosidases previously studied in metazoan fucosidases [25], bifidobacterial fucosidase sequences listed in CAZy reveal substantial in silico differences regarding to their conserved domains, even those ones clustered in the same GH, revealing different adaptation/specialization ranges as well as their origin.
2. Bifidobacterial GH29 Fucosidases
GH29 fucosidases from bifidobacteria listed in CAZy are shown in Table S1. Based on in silico studies concerning conserved domains released by NCBI Conserved Domains Database (CDD), bifidobacterial GH29 fucosidases could be classified into four different phylogenetic groups (Table S1). That differentiation was also confirmed through sequence homology PCA and cluster analyses (Figure 2).
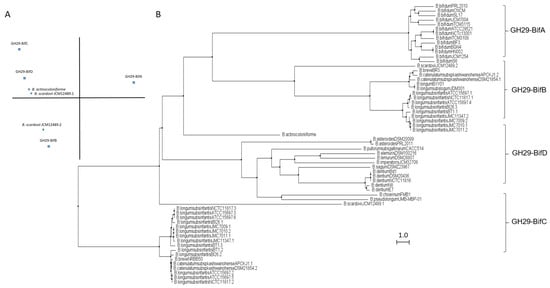
Figure 2. Phylogenetic analysis of bifidobacterial GH29 fucosidases. PCA (A) and cladogram tree (B) distributions of bifidobacterial GH29 fucosidase sequences listed in CAZy, released from Jalview 2.11.1.4 software using the neighbor-joining method.
The enzymes included in the proposed GH29-BifA, only found in B. bifidum strains, are characterized as large membrane-bound fucosidases (AfuC super family domain; NCBI CDD accession number cl34656) and exhibit an accessory F5/F8 type C domain family (NCBI CDD accession number cl23730), probably involved in recognizing galactose or N-acetyllactosamine [26]. Interestingly, while InterPro database (EMBL-EBI) recognized the F5/F8 type C domain (IPR000421), it interpreted the AfuC domain as Glyco_Hydro_29 domain (IPR000933), probably due to the degree of updating of both databases (Table S1). In addition, Ashida et al., 2009 identified a second putative sugar-binding domain in GH29 fucosidase AfcB from B. bifidum JCM1254, domain that is frequently found in membrane-bound or cell-wall-associated proteins and denominated FIVAR [27]. Those results were here confirmed by SOSUI and HMMTOP databases, which allowed the identification of two putative transmembrane helices in GH29-BifA fucosidases (Table S1). Therefore, it has been suggested that both accessory F5/F8 type C and FIVAR domains allow the extracellular character of GH29 fucosidases in B. bifidum and could enhance affinity toward fucosyl conjugates [27]. Moreover, in all the N-terminal regions of GH29-BifA fucosidases, hydrophobic sequences predicted by SignalP-5.0 to be putative signal peptide with potential cleavage sites were observed (Table S1).
Concerning the AfuC/Glyco_hydro_29 domain, the only representative GH29 fucosidase of GH29-BifA purified and characterized, which is AfcB from B. bifidum ATCC 1254, is able to hydrolyze 3-fucosyllactose, Lewis blood group substances (a, b, x, and y types), and lacto-N-fucopentaose II and III. However, the enzyme did not act on glycoconjugates containing α1,2-fucosyl residue or on synthetic pNP-fucose [27].
Supporting the in silico characterization of GH29-BifA fucosidases, several studies confirm the ability of B. bifidum to extracellularly hydrolyze FHMOs [28]. However, B. bifidum appears to prefer the utilization of lactose when growing on FHMO, probably releasing fucose to the environment [28]. This incapacity to consume fucose may be due to the lack of specific transporters. Nevertheless, the extracellular fucosidase activity of B. bifidum could facilitate the establishment of the bifidobacteria community, allowing them to consume the released fucose residues [29].
In contrast to GH29-BifA, the rest of the GH29 fucosidases from bifidobacteria do not have either putative signal peptides or transmembrane helices and consequently their mode of action can be considered intracellular. Indeed, GH29-BifB fucosidases are characterized by exhibiting an AfuC super family/Glyco_Hydro_29 domain (NCBI CDD accession number cl34656/IPR000933) such as GH29-BifA fucosidases but lacking F5/8 type C and FIVAR domains. Due to the presence of the same fucosidase domain in both groups of fucosidases (GH29-BifA and GH29-BifB), similar metabolic capacities could be affirmed. In fact, the only characterized bifidobacterial GH29-BifB fucosidase (Blon_2336 from Bifidobacterium longum subsp. infantis ATCC 15697) revealed similar activity to AfcB from B. bifidum ATCC 1254 (GH29-BifA) against Fuc-α1,3 glucosidic, Fuc-α1,3GlcNAc, and Fuc-α1,4GlcNAc linkages [21]. These GH29-BifB fucosidases appear to be distributed along strains of different species, contrary to GH29-BifA fucosidases, and frequently, strains that exhibit GH29-BifB fucosidases also show GH29-BifC fucosidases, which are duplicated in some of the sequenced strains (Table S1). Actually, the duplication of GH29 fucosidases has been reported previously and plays an important role in fucosidases evolution [30].
GH29-BifC fucosidases are characterized by showing conserved α-Amylase catalytic domain family (NCBI CDD accession number cl38930). It must be taken into account that this superfamily is present in a large number of GHs able to hydrolyze α1,4/6 glycosidic bonds, although in turn they have specific domains unlike the GH29-BifC fucosidases of bifidobacteria [31]. However, since GH29-BifC fucosidases can catalyze the transformation of fucosidic α1-2Gal/3GlcNAc linkages in LNFP I and III, respectively, and mainly Fuc-α1,6 GlcNAc linkages [32], activity non described in the above fucosidase groups, it is difficult to ensure that its catalytic family proposed is α-Amylase catalytic (NCBI CDD) or Glyco_Hydro_29 (InterPro) (Table S1). In this sense, InterPro database (EMBL-EBI) indicated the presence in GH29-BifC fucosidases of a second catalytic family denominated FUC_metazoa_typ (IPR016286) that is close to eukaryotic fucosidases (Table S1). Probably the presence of this domain is key for these fucosidases to be considered as the most unspecific and versatile fucosidases of bifidobacteria since a wide range of substrates has been reported for two different GH29-BifC fucosidases from B. longum subsp. infantis ATCC 15697 [21][27][21,27].
Both GH29-BifB/C fucosidases described in B. longum subsp. infantis strains are likely found in the cytosol. Therefore, efficient transport of oligosaccharides is needed, unlike B. bifidum [13][21][13,21]. In this context, genomic studies carried out on B. longum subsp. infantis ATCC 15697 have unraveled several putative fucose permeases that may facilitate environmental scavenging when soluble fucose is encountered.
In order to elucidate the roles and fitness of the bifidobacterial community to shape the gut microbiome and taking into account the relevance of fucosidases in this regard, their features mentioned above should be updated and expanded to avoid ambiguities in the catalytic domains and relate them to their metabolic properties. Certainly, the rest of the enzymes from different bifidobacterial species need to be characterized in order to reliably distinguish the properties of each group of fucosidases for determining the interaction and mode of actions of bifidobacteria during gut colonization. In this sense, the role of GH29-BifD of fucosidases remains unknown despite having been sequenced and identified in certain Bifidobacterium species (Table S1). Unlike to GH29-BifC, GH29-BifD fucosidases exhibit specific α-L-fucosidase main domain (NCBI CDD accession number cl38930). Surprisingly, their accession number is matching with superfamily AmyAc family of group II, suggesting a better accurate and updated in silico annotation. However, InterPro database (EMBL_EBI) indicates both catalytic domain Glyco_Hydro_29 and FUC_metazoa_typ (InterPro IPR000933 and IPR016286, respectively). Nevertheless, physicochemical properties, substrate specificity confirmation, and their correlation with catalytic domains are still pending to be characterized.
3. Bifidobacterial GH95 Fucosidases
Similar to GH29 bifidobacterial fucosidases and according to architecture domains, bifidobacterial GH95 fucosidases collected on CAZy could also be subclassified into two main groups (Table S2; Figure 3).

Figure 3. Phylogenetic analysis of bifidobacterial GH95 fucosidases. PCA (A) and cladogram tree (B) distributions of bifidobacterial GH95 fucosidase sequences listed in CAZy, released from Jalview 2.11.1.4 software using the neighbor-joining method.
The extracellular character observed in GH29-BifA fucosidases from B. bifidum strains is also reflected in their GH95 fucosidases, which are characterized by a putative signal peptide and two predicted transmembrane helices. Among GH95 fucosidases, those features are only found in the proposed GH95-BifA fucosidases from B. bifidum with the exception of Bifidobacterium saguini DSMZ 23967 fucosidase (Genbank QTB91571.1), which exhibited two putative transmembrane helices (Table S2).
The proposed GH95-BifA was characterized according to the NCBI CDD database by exhibiting Glycosyl hydrolase 65 N-terminal (accession number cl22392) as main catalytic domain, while InterPro database analysis (EMBL-EBI) revealed a Glycosyl hydrolase 95 N-terminal (IPR027414) (Table S2). The observed ambiguous prediction on the catalytic architecture could be due to the lack of updating and mismatch annotations. Nevertheless, a common evolutionary origin for GH65 and GH95 families, among others, with conservation of their putative catalytic amino acid residues, was noticed and likely influenced the in silico results [18]. Nevertheless, and contrary to GH65 family, the only GH95-BifA representative fucosidase recombinantly produced and characterized (AfcA from B. bifidum JCM1254) showed great activity against Fuc-α1,2 Gal linkages, mainly hydrolyzing 2′-Fucosyllactose and lacto-N-fucopentaose I [17][33][17,33].
On the other hand, while NCBI CDD database detected two YjdB overlapping domains (accession number cl35007), whose functions are still uncharacterized but in turn contain Ig-like domain, InterPro database noticed Ig-like_Bact and Bacterial Ig-like group 2 (BIG2) domains instead (accession number IPR022038 and IPR003343, respectively) (Table S2). Despite this coincidence, only the position of one domain practically matches in both databases (YjdB and BIG2) (Table S2). In addition, InterPro identifies Ig-like_Bact near to N-terminal unlike NCBI CDD, and probably GH95-BifA sequences could exhibit up to three accessory domains.