Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Camila Xu and Version 1 by Andrea Olmos-Ortiz.
Gestational Diabetes Mellitus (GDM) is a transient condition characterized by carbohydrate intolerance, hyperglycemia, peripheral insulin resistance, insufficient insulin secretion or activity, endothelial dysfunction, and low-grade inflammation during pregnancy, frequently with the first onset between 24 and 28 weeks of gestation.
- inflammation
- cytokines
- adipokines
- antimicrobial peptides
- oxidative stress
- metabolic stress
1. Introduction
Gestational Diabetes Mellitus (GDM) is a transient condition characterized by carbohydrate intolerance, hyperglycemia, peripheral insulin resistance, insufficient insulin secretion or activity, endothelial dysfunction, and low-grade inflammation during pregnancy, frequently with the first onset between 24 and 28 weeks of gestation [1]. Although it is a transient, GDM effects can last beyond the perinatal period and impact the health of mother and fetus in both short- and long-term [2,3,4][2][3][4].
In 2017, it was estimated that 21.3 million births (16.2%) worldwide were affected by hyperglycemia during pregnancy, with GDM contributing 86.4% of these cases [5,6][5][6]. Furthermore, an increase in the prevalence of GDM effects is expected due to the parallel increasing rate of pre-gestational obesity and excessive weight gain during pregnancy.
It is well accepted that a key event in the onset of GDM is the maternal peripheral insulin resistance. During normal pregnancy, there is a transient and physiological state of decreased insulin sensitivity, necessary to prioritize fetal glucose uptake. In response, β-cells proliferate and synthesize more insulin as a mechanism to counteract insulin resistance and favor euglycemia. However, before pregnancy, some women have a first pancreatic hit by GDM risk factors such as pre-gestational overweight, obesity, hypercaloric diet, personal or familiar antecedent of GDM, advanced maternal age, or presence of insulin resistance disorders such as polycystic ovarian syndrome [1,7][1][7]. With pregnancy, these women are exposed to a second pancreatic hit: the insulin resistance associated with early pregnancy. These two hits lead to GDM development because of inadequate compensatory changes in β-cell mass activity and proliferation due to a more pronounced insulin resistance condition, particularly during the second and third trimester of pregnancy [8,9][8][9]. This transient metabolic stress on the pancreas during pregnancy, partially explains why GDM is associated with a higher risk of post-partum development of Type 2 Diabetes Mellitus (T2DM) in the mother and fetus [10].
The American College of Obstetricians and Gynecologists (ACOG) recognizes two types of GDM. GDM Class 1 (A1GDM) patients respond to diet intervention (low glycemic index meals with low simple sugar and high fiber content) and exercise. GDM Class 2 (A2GDM) patients need pharmacologic treatment to achieve target glucose levels. For A2GDM, the first-line therapy recommended by the American Diabetes Association (ADA) is insulin, preferentially short-acting insulin (i.e., Lispro or Aspart), and long-acting insulin (i.e., Glargine or Detemir) [7,11][7][11]. However, other Societies including ACOG, German Diabetes Association, German Society of Gynecology and Obstetrics, and The Society for Maternal-Fetal Medicine recommend metformin instead [1,12][1][12]. Recent studies showed a lower risk for preeclampsia, macrosomia, neonatal hypoglycemia, and hypertensive disorders as well as better outcomes in maternal weight gain and glycemic control. No difference was observed in rates of caesarean section, neonatal respiratory distress and preterm birth compared to insulin treatment alone [13,14,15,16,17][13][14][15][16][17]. There is insufficient evidence on the long-term effects of prenatal exposure to metformin (especially because it crosses the placenta). Two recent studies showed no difference in growth and development in children of metformin-treated and insulin-treated mothers over a four-year period [18,19][18][19]. More long-term studies are needed to understand the long-term effects of metformin during pregnancy.
As it would be expected, hyperglycemia and GDM disturb placental ultrastructure and morphophysiology since the early stages of the disease. Reported placental abnormalities in GDM patients include increased placental weight, intimate glycogen deposits, increased number of syncytial knots, villous edema, and larger syncytial area and volume for favoring nutrient uptake [20,21,22,23][20][21][22][23]. Histopathologic analysis of GDM placentae indicates enhanced angiogenesis and high vasculogenesis rate evidenced by increased villous vascularity often associated with thickened immature villi capillaries and signs of placental hypoperfusion [24,25][24][25]. There has also been reported increased fibrinoid necrosis, chorangiosis, and ischemia [25,26][25][26]. Additionally, GDM syncytiotrophoblasts present an exaggerated mitochondrial dysfunction accompanied by a lower rate of glycolysis, oxidative phosphorylation, and ATP synthesis, which indicates a compromised metabolic supply and therefore placental overstress [27,28][27][28]. Lipid metabolism of the placenta is also distorted in GDM with evidence of larger lipid droplets, higher triglyceride accumulation, and fatty acid transporter expression [28,29][28][29]. This is all indicative of modification of the endocrine, immune, angiogenic, and antioxidant functions by placentae in GDM mothers.
2. Role of Placenta in the Endocrine Milieu of GDM
The endocrine system is the earliest system developing during intrauterine life. From the stage of two-blastomeres, the embryo begins to secrete the beta-fraction of the human chorionic gonadotrophin (β-hCG), which has been suggested to be a product of mRNAs previously stored in oocytes [30,31][30][31]. Later, by 6 days post-fertilization, the trophectoderm establishes its endocrine phenotype through the de novo synthesis of β-hCG. The early placenta then turns on its hormonal switch and maintains secretion of a broad panel of hormones with central activities in the maintenance of pregnancy, fetal growth, and development. These include somatostatin, placental lactogens, placental growth hormone (PlGF), gonadotropin-releasing hormone, corticotropin-releasing hormone, thyrotropin-releasing hormone, progesterone, and estradiol besides other growth factors such as the Insulin-like growth factor 1 and 2 (IGF-I and IGF-II) [32].
Inherent to its secretory phenotype, the placenta is also a target of hormonal biological activity because it expresses most receptors for these hormones and growth factors. Therefore, placental hormones act in endocrine, paracrine, and autocrine pathways in the Maternal-placental-fetal-unit (MPFU).
In the context of GDM, diverse hormonal actors take part in the beginning or progression of the hyperglycemic, insulin resistance, oxidative stress, and the meta-inflammatory state of this disease. Three years ago, Madhusmita Rout and Sajitha Lulu proposed a network of maternal and placental genes and their potential impact on the transport of nutrients from mothers with GDM to their babies [33]. Through diverse in silico tools analyzing the interaction of gene/protein/miRNA/transcription factors, they described an important dysregulation of certain placental hormones, including leptin, insulin/IGF-I and their receptors, and the placental growth hormone receptor. Recently, other hormonal candidates that might play a role in the pathophysiology of GDM have been described through comprehensive bioinformatics, gene analysis, ROC analysis, and RT-PCR, including β-hCG, oxytocin receptors, binding proteins of IGF-I, and some cytochromes involved in the steroidogenic pathway [34,35][34][35].
2.1. The Insulin and IGF-I/IGF-II Axis and Molecular Pathways Primarily Disturbed in GDM Placentae
Insulin resistance is one of the first alterations in the pathogenesis of GDM. Insulin acts through two known receptor isoforms: insulin receptor A (IR-A) and insulin receptor B (IR-B). These are heterodimer receptors composed of an extracellular α-subunit that binds to insulin, and an intracellular β-subunit that binds to the insulin receptor substrate 1 (IRS-1). Both isoforms are transcripts of the same INS gene. However, the IR-A product lacks a sequence of 36 nucleotides in the C-terminal of the α-subunit as a result of alternative splicing of exon 11. In contrast, IR-B is the result of the full transcription of the INS gene [36,37][36][37]; this explains the differences in their activities. Insulin has a higher affinity for IR-A than for IR-B. In addition, IR-A activates the Ras-Raf-mitogen-activated protein kinase (MAPK) cascade, whereas IR-B activates the phosphatidylinositol 3-kinase (PI3K) and protein kinase B (Akt) signaling.
The study of the cellular pathways activated by insulin began around 33 years ago [38]. Since then, diverse research papers have scrutinized these cellular cascades, and finally, in 1995, the role of PI3K and Akt in the glycemic control and other metabolic activities of insulin was described [39,40][39][40]. After IR-B binding to insulin, PI3K binds to tyrosine-phosphorylated IRS proteins, leading to the formation of phosphatidylinositol (3,4,5)-triphosphate (PIP3). Downstream effects of PIP3 lead to activation of 3-phosphoinositide dependent protein kinase (PDK)1 and the subsequent activation of a variety of kinases, of which Akt 1–3 is the best-studied [41]. Main metabolic activities of insulin are related to Akt phosphorylation: (i) increased glycogen synthesis by inactivation of glycogen synthase kinase-3 (GSK3) α/β and activation of glycogen synthase [39]; (ii) Decreased transcription of gluconeogenic genes in liver and autophagy genes in muscle, by phosphorylation of forkhead box (FOX) transcription factors [42]; (iii) stimulated protein synthesis and suppression of autophagy by phosphorylation of tuberous sclerosis 2 (TSC2) and the 40 kDa proline-rich Akt substrate (PRAS40) which leads to activation of mTORC1 [43,44][43][44]; (iv) increased glucose uptake by phosphorylation of TBC1 domain family member 1/Akt substrate of 160 kDa (TBC1D4/AS160) which regulates trafficking and translocation of GLUT4 cytoplasmic vesicles to the plasma membrane [45]. In line with their metabolic effects, IR-B is preferentially expressed by classical insulin-sensitive cells such as hepatocytes, adipocytes, and myocytes, which have important roles in glucose, lipid, and protein metabolism.
On the other hand, activation of IR-A induces the Grb-2/Erk 1/2 MAPK pathway related to cell growth, differentiation, and survival processes [46]. This isoform is predominantly expressed in cancer tissues, the brain, hematopoietic cells, and the placenta [36].
Although the human placenta expresses both insulin receptors, IR-A is expressed at higher levels than IR-B [47]. This differential ratio is probably related to the need for tight control of the crucial proliferative and pro-differentiation pathways during pregnancy. On the other hand, there are redundant placental pathways to help in the vital fetoplacental glucose transfer, besides insulin/IR-B mediated GLUT-4 translocation [48[48][49],49], as occurs in insulin-dependent tissues. During the first trimester of pregnancy, IR-A is expressed more in the apical membrane of syncytiotrophoblasts whereas at term IR-B is concentrated in endothelial cells of the villi microvasculature [50].
In Figure 1, we summarize the disturbed molecular pathways in GDM placentae. Immunohistochemical and blotting studies showed that GDM placentae have a lower total protein expression of IR-A, PIP3, and IRS-1 in comparison to placentae from uncomplicated pregnancies. Interestingly this occurs independently of the metabolic control of the disease which implies profound and sustained effects in placenta signaling networks [51,52][51][52]. Also, it seems that the obesogenic environment is an additional regulating factor of insulin signaling among GDM placentae, involving IRS-2, PI3K, and GLUT4, the most sensitive targets to obesity [53]. Additionally, it has also been reported that GDM placentae have a pronounced phosphorylated pattern of IR and IRS-1 proteins, accompanied by hyperphosphorylation of STAT-3, MAPK 1-3 (Erk 1/2), and Akt [54,55][54][55]. Altogether, these data suggest that GDM primes the placenta to overstimulate the insulin signaling to compensate sustained exposure to hyperglycemia. The deficit in the total protein levels of these mediators results in insufficient placental glucose uptake, and consequently in a hyperglycemic state [56,57][56][57]. Although, other authors did not observe differences in the total protein levels of placental IR and IRS-1 [54,58][54][58]. Considering these inconsistencies, we believe more studies are needed to clarify insulin signaling in GDM placentae and to understand how placental imbalance in these signaling pathways results in higher levels of inflammatory cytokines, adipokines and oxidative reactive species, insulin resistance, and vascular disorders, all of which prevail in the local placenta and peripheral tissues of GDM mothers.
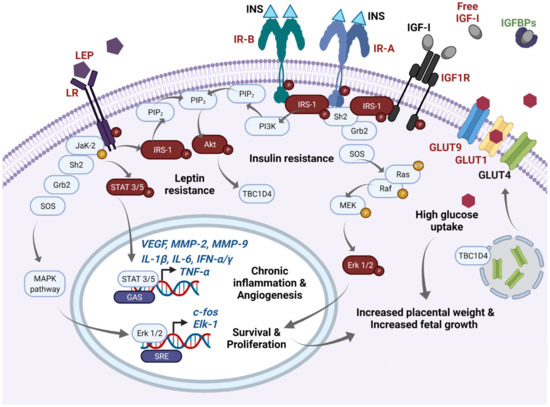
Figure 1. Main molecular pathways disturbed in GDM placentae. In this schematic, we highlight in red main molecules reported as overexpressed/overactivated in GDM placentae, whereas downregulation is highlighted in green. High expression of phospho-IR-A and phospho-IR-B has been reported in the GDM placenta. After insulin binding, IR-A phosphorylates IRS-1 and recruits Scheme 2. and Grb2 proteins which induce the GTPase activity of Ras, and then a GDP is exchanged by a GTP. This initiates a subsequent cascade of phosphorylations of Raf, MEK, and Erk 1/2. Finally, overexpressed phospho-Erk proteins translocate to the nucleus and recognize SRE sites favoring active transcription of c-fos and Elk-1. FOS proteins have been implicated as regulators of cell proliferation, survival, differentiation, and transformation. This pathway mediates increased placental weight and increased fetal growth in GDM. Additionally, increased levels of free IGF-I resulting from low IGFBPs serum concentrations also activate IR-A signaling as well as IGF1R and are related to proliferative effects. IGF-I excess has been also implicated in macrosomia and excessive placental growth in GDM women. On the other hand, after insulin binding to IR-B, phospho-IRS-1 activates PI3K and leads the formation of PIP3 from PIP2. Then, PIP3 activates Akt which mediates diverse metabolic effects; one of them includes GLUT4 translocation from endosomes to the cellular membrane through TBC1D4 signaling. High expression of GLUT1 and GLUT9, and probably GLUT4, mediates high glucose uptake in the GDM placenta, which can also participate in fetal and placental growth through an excess of this energetic substrate. Finally, GDM placentae present high expression of leptin and its receptor. High expression of phospho-leptin receptor recruits JaK-2 protein and activates STAT 3 or 5 proteins. Then, STATs dimerize and translocate to the nucleus and recognize GAS sites and provoke transcription of VEGF, MMP-2, MMP-9, TNF-α, IL-1α, IL-1β, IFN-α, IFN-γ, among others, which exacerbate the inflammatory placental milieu and contribute to stimulating the angiogenic process. Additionally, activation of the leptin receptor can also crosstalk with MAPK and Akt pathways. Sustained overactivation of all these pathways finally leads to clinical insulin and leptin resistance. GAS: Gamma-activated sequence. GDM: Gestational diabetes mellitus; GLUTs: Glucose transporters; IGFBPs: IGF-I binding proteins; IGF-I: Insulin-like Growth Factor 1; IGF1R: IGF-I receptor; INS: Insulin; IR-A: Insulin receptor type A; IR-B: Insulin receptor type B; LEP: Leptin; LR: Leptin receptor; SRE: Serum response elements.
Another system that can cross-react with insulin signaling is the IGF-I axis. The system consists of two ligands IGF-I and IGF-II, two receptors IGF-1R and IGF-2R, six IGF binding proteins (IGFBP 1–6), and four insulin-like growth factor binding protein-related peptides IGFBP-rP1-4 [55,59][55][59]. These growth factors mainly regulate growth and metabolism throughout the life cycle, but its activity is critical during intrauterine life for mammalian development. They provide a signal to cells to indicate that adequate nutrients are available, and therefore to enhance cellular protein synthesis, to favor hypertrophy, and to stimulate cell division [60].
Insulin and IGF-I present a relatively low homology of 34% (BLAST alignment: CAA40342.1 and AAN39451.1), but their receptors are highly homologous, around 57% (BLAST alignment: CAA28030.1 and AAA59452.1). These similarities in their structures generate promiscuous interactions between them. IGF-II binds to IR-A with an affinity close to that of insulin, but it does not bind to IR-B [61]. Additionally, random hybrids of IR-A/IR-B and hybrids of IRs/IGF-1R have been reported in the placenta [36,62,63][36][62][63]. IGF-1R/IR hybrids bind IGF-I and IGF-II with high affinity but bind insulin with a relatively low affinity [64].
Given the structural similarities between components of the IGFs/Insulin axis, it is not surprising that these hormones share signaling pathways. IGF-I binds to IGF-1R and activates two main cascades: (i) PI3K/Akt pathway via IRS-1 phosphorylation which predominantly leads to metabolic effects; (ii) Ras-Raf-MAPK pathway via SHC domain proteins which control cellular growth and differentiation [59]. On the other hand, IGF-II binds with high affinity to IGF-2R, also known as the cation-independent mannose 6 phosphate receptor. This interaction targets IGF-II for its lysosomal degradation and consequently, IGF-2R sequesters IGF-II, controlling the circulating levels of this hormone. Therefore, the biological activity of IGF-II is exclusively derived from its binding to IR-A or IGF-1R, considering that it does not bind to IR-B, as mentioned before [65].
The placenta synthesizes all components of the IGF axis from early stages at 7 weeks of gestation. IGF-I was more expressed in the second and third trimesters of pregnancy in comparison with early pregnancy, and it was expressed by practically all placental cells except syncytiotrophoblasts. Whereas IGF-II was profusely expressed by cytotrophoblasts, mesoderm core, basal plate, columnar cytotrophoblasts, amnion, and chorion. IGF-IR was expressed ubiquitously in the placenta, except in Hofbauer cells. All six IGFBPs were expressed in decidua basalis and parietalis [66].
One key insulin-like action of IGF-I is related to glucose metabolism [67]. Biomedical and clinical studies indicate that IGF-I is a hypoglycemic factor that increases glucose uptake in different kinds of cells, including euglycemic trophoblasts [68,69,70][68][69][70]. However, deeper studies are needed to confirm its metabolic effects in the GDM milieu. In contrast, IGF-II seems to present a hyperglycemic effect since overexpression of IGF-II in pancreatic β-cells results in the development of T2DM [71].
Excessive fetal growth and weight is a common complication from GDM newborn babies. Macrosomia has been explained by two central modulators: hyperglycemia and activation of the IGF-I axis. Maternal hyperglycemia increases energetic substrate availability and then stimulates excessive growth and adiposity in GDM mothers [72]. In fact, an increased concentration of glucose transporters GLUT1 and GLUT9 has been observed in GDM placentae, which favors an increased placental and fetal D-glucose uptake [49,52,73,74][49][52][73][74] (see Figure 1).
Concerning IGF-I signaling, different serum components of the pathway have been measured in GDM patients in the last 20 years. In a recent meta-analysis developed by Dr. Wang’s group, in which they analyzed 12 independent studies, they found GDM was consistently associated with higher maternal IGF-I levels in mid-gestation (20–29 weeks) and late-gestation (>30 weeks), whereas serum IGF-II did not present significant changes between GDM and control mothers [60]. Interestingly, most data show significantly lower cord levels of IGFBP1, IGFBP2, IGFBP3, IGFBP-6 or IGFBP-rP1 [55[55][75][76],75,76], and lower maternal levels of IGFBP1 and IGFBP2 [55]. All IGFBPs bind both IGF-I and IGF-II with similar affinities (except IGFBP-6 which is essentially IGF-II specific [77]). Their metabolic effects are related to inhibition of IGF-I signaling by sequestrating it into a circulation reservoir. Consequently, diminished levels of IGFBPs and IGFBP-rPs result in higher cord blood levels of free-IGF-I in GDM patients [75]. Further, the research group of Dr. Sciacca identified an increased phosphorylation pattern of IGF-1R in placentae from metabolic uncontrolled mothers with GDM and T2DM [58]. These changes support a persistently activated IGF-I signaling in GDM placentae by increased activity of free-IGF-I (see Figure 1).
It is well known that the growth hormone (GH)-IGF-I axis is the major regulator of longitudinal growth along life. In addition, significant and positive correlations between the birth weight of newborns from GDM mothers and maternal serum IGF-I or molecules of the IGF-I signaling in GDM placentae have been described [52,78,79][52][78][79]. Therefore, overactivation of IGF-I signaling may be one critical factor involved in the development of macrosomia in babies from GDM mothers. Other morphologic changes in the placenta are also related to macrosomia, including broader intervillous spaces, increased terminal villus volume, a large proportion of immature villi, and a larger syncytiotrophoblast surface allowing higher amounts of glucose to cross the placenta [20,21][20][21].
One additional hypothesis in excessive fetal weight gain in GDM is related to IR/IGF-1R hybrids. A high proportion of these hybrids has been reported in skeletal muscle and adipose tissue of T2DM patients [63,80[63][80][81],81], and in placentae from insulin-resistant women [82]. IR/IGF-1R hybrids increase the binding sites for IGF-I and IGF-II, favoring IGF signaling, proliferation, and anabolic processes, as has been previously published in cancer models [83]. However, this hypothesis, which deserves to be further explored, has not so far been studied in the placentae of mothers with GDM.
2.2. The Role of Pancreatic β-Cells and Lactotroph Hormones in GDM
In a healthy pregnancy, insulin resistance increases between 50% and 60% in the third trimester, compared to the pre-pregnancy period [84]. This physiologic resistance is needed to ensure adequate delivery of glucose to a fast-growing fetus. In response, the mother needs to expand her capacity for insulin secretion which is achieved by an increase in β-pancreatic cell mass and number, finally leading to a euglycemic pregnancy. The lactotroph hormones prolactin (PRL) and human placental lactogens participate in cell-specific β-cell responses to counteract the physiological insulin resistance developed during pregnancy. These pancreatic adaptations occur before the onset of insulin resistance in pregnancy [9].
The maternal decidua is the main extra-pituitary source of PRL synthesis [85[85][86],86], although columnar trophoblasts and villous cytotrophoblasts of the placenta can also synthesize it to a lesser extent [85,87][85][87]. On the other hand, placental lactogens are exclusively synthesized during pregnancy by fetal syncytiotrophoblasts and include human placental lactogen (hPL), human chorionic somatomammotropin A and B (hCS-A and hCS-B), and the placental growth hormone (PGH). Evolutionary studies indicate that placental lactogens are closely related in their chemical structure to the human Growth Hormone (GH) as a result of three duplications and one deletion in the GH gene [88]. Derived from this structural homology, lactogens share with GH their binding capacity to both somatogenic and lactogenic receptors [89]. GH binds primordially to the hGH receptor and acts as a somatogen, whereas PRL and hPL bind to the prolactin receptor (PRL-R) and act as lactogens. PRL-R is a member of the cytokine receptor superfamily which presents 3 structural regions: an extracellular ligand-binding domain, a hydrophobic transmembrane domain, and an intracellular signaling domain. Multiple promoters and alternative splicing of the PRLR gene generate several isoforms which vary exclusively in their intracellular domains and potential recruitment of signaling mediators [90]. After ligand binding, PRL-R dimerizes which leads to the trans-phosphorylation of tyrosine residues present in Janus kinase 2 (JaK-2). This is followed by recruitment of signal transducers and transcription activators (STATs) -1, -3, or -5 which dimerize and migrate to the nucleus to enhance the expression of PRL-dependent genes [91]. During pregnancy, the binding of hPL or PRL to the long isoform of PRL-R in pancreatic β-cells activates the JaK-2/STAT-5 pathway which results in metabolic adaptations of these cells characterized for higher transcription of GLUT-2, glucokinase, insulin, survivin, cyclin D2, and Bcl6 genes [92]. GLUT-2 favors glucose uptake by β-cells, then glucose is phosphorylated by glucokinase and enters glycolysis/Krebs cycle/oxidative phosphorylation to increase ATP production. Higher ATP/ADP ratio blocks ATP-sensitive potassium channels, K+ accumulation depolarizes β-cells, and voltage-gated calcium channels become activated. The resultant rise in intracellular Ca2+ triggers insulin secretion [93]. Additionally, transcription of survivin, cyclin D2 and Bcl6 genes increases cell mitotic divisions, avoids apoptosis, and stimulates the expansion of pancreatic islets during normal pregnancy [94]. Beta-cell proliferation is also dependent on the downstream serotoninergic effect of both, PRL and hPL [95]. hPL is more potent than PRL to increase insulin secretion and β-cells proliferation, whereas GH has lower potency [96,97][96][97]. We now know that the morphologic changes in pancreatic β-cells related to pregnancy occurs largely through hPL and PRL action, but Hepatic Growth Factor (HGF), Epidermal Growth Factor (EGF), vitamin D, progestins and estrogens are also implicated [98,99,100,101][98][99][100][101] (see Figure 2).
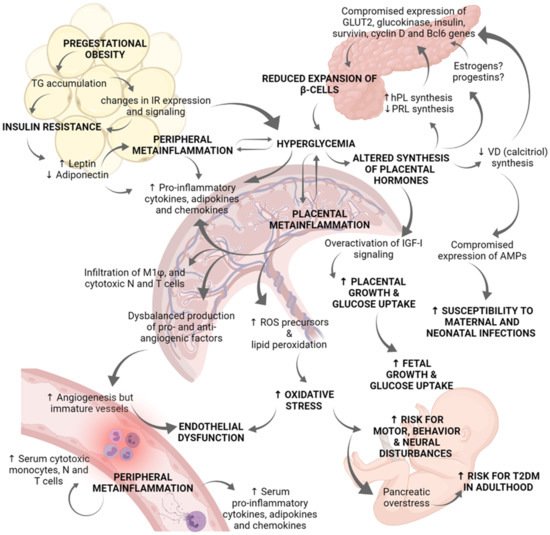
Figure 2. Role of placenta in the immunoendocrine dysregulations occurring in gestational diabetes mellitus. Hyperglycemia during pregnancy compromises the correct physiology of diverse organs, and particularly the placenta. Pregestational obesity is characterized by an excessive TG accumulation and changes in IR expression and signaling in adipose tissue, as well as in skeletal muscle cells. These changes in insulin-dependent tissues lead to insulin resistance. Then, adipose tissue secretes high levels of leptin and inhibits those of adiponectin. Leptin activation of JaK-2/STAT 3/5 pathway results in increased cytokine production contributing to peripheral metainflammation. Due to insulin resistance and altered IR signaling, women suffer chronic hyperglycemia. Clinical and biomedical studies indicate there is a positive regulatory loop between hyperglycemia and metainflammation, in which hyperglycemia induces placental and peripheral synthesis of pro-inflammatory cytokines, chemokines, and adipokines. Metainflammation also alters GLUT and IR expression which worsen hyperglycemia status. The synthesis of placental hormones is altered by hyperglycemia. Deficient synthesis of PRL and calcitriol (the active form of vitamin D) has been reported in GDM placentae, whereas hPL synthesis is increased. These placental hormonal changes, and probably estrogens and progestins, compromise pancreatic gene expression of GLUT2, glucokinase, insulin, survivin, cyclin D2, and Bcl6; all these genes are related to b-cells proliferation and survival. Because of reduced b-cells expansion, hyperglycemia worsens. Additionally, deficient synthesis of calcitriol abates placental expression of antimicrobial peptides related to innate defense, increasing mother and fetus vulnerability to infections in the perinatal period. Another endocrine dysregulation is derived from IGF-I overactivation in the placenta, which explains increased placental growth and glucose uptake. The establishment of a chronic inflammatory milieu in the placenta results in: (i) Increased production of pro-inflammatory cytokines, chemokines, and adipokines; (ii) Increased placental infiltration of M1 macrophages, cytotoxic neutrophils, and T cells; (iii) Deregulated production of pro-angiogenic and anti-angiogenic factors; and (iv) Increased lipid peroxidation and synthesis of ROS precursors. Even if angiogenesis is induced, vessels in the placenta are thickened and immature. Overall, altered vessels formation, hyperglycemia, and oxidative stress induce endothelial dysfunction. Furthermore, the serum inflammatory profile is evidenced by high levels of pro-inflammatory cytokines, chemokines, and adipokines as well as a higher presence of cytotoxic monocytes, neutrophils, and T cells. Hyperglycemia and over-activation of IGF-I signaling results in increased fetal growth which may contribute to macrosomia. Finally, hyperglycemia and oxidative stress produce pancreatic overstress in the fetus, causing an increased risk for T2DM development later in life. Experimental and clinical evidence indicates that GDM fetuses present a marked neural pro-oxidative environment which may lead to neural, motor and behavior disturbances. AMPs: antimicrobial peptides; GDM: Gestational diabetes mellitus; GLUT: Glucose transporter; hPL: human placental lactogen; IGF-I: Insulin-like Growth Factor 1; IR: Insulin receptor; N: Neutrophils; PRL: Prolactin; ROS: Reactive oxygen species; T: T lymphocytes; T2DM: type 2 Diabetes mellitus; TG: triglycerides; VD: vitamin D.
Pregnancy is considered a physiologically hyperprolactinemic state, in where PRL potentiates glucose-stimulated insulin secretion and β-cell mass. However, exacerbated hyperprolactinemia, as occurs with a prolactinoma, is related to insulin resistance [102]. In GDM, blood levels of hPL are higher than in normal pregnancies and correlate with increased placental weight, macrosomia, hyperglycemia, insulin resistance, and altered values in an Oral Glucose Tolerance Test (OGTT) [103,104][103][104]. In contrast, GDM mothers present similar or even lower levels of PRL than normal pregnant women [105]. This relative contradiction seems to indicate that a delicate balance of PRL and hPL during pregnancy is needed to achieve adequate pancreatic β-cells proliferation and to avoid insulin resistance [106]. In a diabetic mouse model, low- and high- PRL treatment induced β-cell proliferation; however, low PRL levels reduced hepatic insulin resistance whereas high PRL exacerbated it and elevated apoptosis of β-cells [107]. A DNA sequencing study also supports the critical role of PRL-R in the control of glucose metabolism in GDM patients. In particular, two single nucleotide polymorphisms in the PRLR gene were associated with a 2-fold risk for developing GDM [103]. The study of physiological control of pancreatic β-mass expansion by lactotroph hormones still needs to be enlarged in the context of GDM and their co-interactions with estrogens/progestins hormones, inflammation, and obesity.
3. Hyperglycemia-Induced Metainflammation in GDM Alters Placental Immune Cells Population Favoring an Inflammatory Cytokine Signature and an Imbalance in Adipokines and Defense Peptides
The persistent exposure to hyperglycemia in GDM mothers results in a systemic response of inflammation named metainflammation [108,109][108][109]. This term defines a sustained low-grade inflammatory state characterized by an increase in serum levels of pro-inflammatory cytokines and tissue macrophage infiltration in the absence of tissue damage. Metainflammation in the context of GDM is favored by metabolic disorders such as maternal obesity or excessive weight gain, which induce inflammatory pathways leading to insulin resistance [110]. This persistent inflammatory state in GDM mothers has been associated with an increased risk for diabetes, obesity, and other poor outcomes and diseases in their offspring [111,112,113,114][111][112][113][114].
Several clinical studies in GDM patients have demonstrated alterations in a broad profile of inflammatory mediators, including lower serum levels of anti-inflammatory interleukin (IL)-10 and adiponectin, higher serum levels of pro-inflammatory TNF-α and IL-6, increased maternal serum adipokines (chemerin, leptin, omentin, visfatin, and the fatty acid-binding protein 4 FABP-4), Th1 cytokines (INF-γ, IL-2, IL-18) and chemokines (CXCL16, IL-8) [115,116,117,118,119,120][115][116][117][118][119][120]. Intracellular signaling of these inflammatory mediators is related to a worse fetal prognosis, such as preterm delivery and premature rupture of membranes [121].
3.1. The Role of Immune Cells in GDM Placentae
It is well known that hyperglycemia affects innate immunity and that there is a diabetes mellitus-dependent vulnerability to infections [122]. This scenario affects the correct function of innate immune cells as well as Toll-like receptor (TLR)–dependent responses at the MPFU, including alterations in the immune functions of monocytes/macrophages, dendritic cells, NK cells, granulocytes, and Hofbauer cells.
Similar to what is seen in adipose tissue, obesity and GDM predispose to macrophage, granulocyte, and T lymphocyte infiltration into the placenta [120,123,124][120][123][124]. Indeed, the placenta expresses mRNA for CD68, CD14, EMR-1, and TCRa (immune cell infiltration markers), which are known to be increased in GDM placentae [123,124][123][124]. Macrophages infiltrated in GDM placentae present an M1 phenotype with a strong inflammatory response characterized by high expression of IL-6, TNF-α, IL-1β, IL-8, and the monocyte chemoattractant protein 1 (MCP-1) [120,123][120][123].
Hofbauer cells are fetal macrophages immersed in placental villous and present an anti-inflammatory profile (M2). In a rat model of GDM, Hofbauer cells switched their M2 profile towards M1 (pro-inflammatory) and induced the oxidative stress pathway [125]. In this regard, Hofbauer cells when treated with high glucose switch their profile to an M1-type, triggering inflammatory pathways [125], similarly to macrophages in a high-glucose environment [126]. However, in human placenta from GDM women, Hofbauer cells seem to preserve their M2 phenotype despite a hyperglycemic environment [127]. M2 macrophages play a relevant role in tissue remodeling during placental development, so further studies are needed to understand the effect of GDM on these cells.
Neutrophil activity is also altered in GDM. Neutrophils seem to be significantly activated, forming a high number of neutrophil extracellular traps (NETs). Under normal circumstances, these web-like structures represent an additional mechanism of the innate immune system to protect us from invading microorganisms. However, in pathological conditions such as diabetes and cancer, platelets may also get trapped, contributing to the pathological effects of NETs, which include damage to the endothelium and thrombotic events. Notably, numerous neutrophil infiltrates have been detected in the placentae of GDM-patients [128]. In addition, elevated neutrophil-derived products including nucleosomes, neutrophil elastase, and free DNA have been found in the plasma of diabetes mellitus patients [129]. Elevated first-trimester neutrophil count has also been associated with the development of GDM and adverse pregnancy outcomes [130]. In the placenta, extracellular traps formation may be triggered by infection (e.g., by bacteria or their products) or by inflammation (e.g., preeclampsia and GDM). Notably, extracellular traps comprise a vast array of molecules with antimicrobial activity, such as elastase, cathepsin G, defensins, myeloperoxidase, hCTD, and bacterial permeability-increasing protein, which explain their bactericidal effect [131]. However, the involvement of these immune structures in some noninfectious, autoimmune, and inflammatory processes grants further studies to understand their participation in GDM pathophysiology.
Regarding NK cells, diverse comprehensive reviews have described the critical participation of NK cells, particularly decidual NK cells with CD56bright/superbright and CD16- phenotype, in pregnancy development [132,133][132][133]. However, few original articles have revised the role of these cells in GDM pathophysiology. A higher percentage of cytotoxic NK cells (CD16+ CD56dim) was observed in maternal serum of overweight GDM patients and placental extravillous tissue in comparison with euglycemic women [119,134][119][134].
3.2. Major Placental Cytokines in GDM
The placenta presents not only an active endocrine function, but also an important immune-modulatory action characterized by the synthesis of diverse cytokines, chemokines, and adipokines, as well as their receptors. The implication of pro-inflammatory cytokines in GDM pathology has been demonstrated in numerous studies [135,136,137,138][135][136][137][138]. Gene microarray experiments in GDM placentae showed increased expression of genes for stress-activated and inflammatory responses, with upregulation of interleukins, leptin, and TNF-α receptors and their downstream molecular adaptors [139].
Undoubtedly, the signaling of NFκB is the main regulator of inflammatory pathways in normal and GDM placentae. After TNF-α binding with their receptor TNFR1, the adaptor protein TRADD is recruited and associated with the death domain of TNFR1. TRADD acts like platform binding for TRAF2 and RIP adaptor proteins which eventually activate the TAK1 kinase to phosphorylate and activate the IKK complex formed by the catalytic subunit IKKα and IKKβ, and the regulatory subunit NEMO. The IKK complex phosphorylates the IκB proteins that are constitutively bound to NFκB, keeping this factor in the cytosol. The serine phosphorylation of IκB proteins promotes their ubiquitination and proteolytic degradation by the proteasome, free allowing the nuclear translocation of NFκB [140,141][140][141]. The NFκB upregulates target genes that encoded pro-inflammatory cytokines, inducing a chronic inflammatory loop that contributes to the development of insulin resistance.
NFκB can be activated by endogenous molecules released during tissue damage and oxidative stress, including debris from apoptotic, saturated fatty acids, heat shock proteins, advanced glycation products (AGEs) which are recognized by the TLR-4 receptor [142]. In GDM and maternal hyperglycemia there is a positive association with an increase of TLR-4 and NFκB signaling in the placenta [143,144][143][144]. TLR-4-induction of NFκB signaling in the placenta is an important mechanism that is altered during gestational diabetes; however, further studies are needed to elucidate the involvement of innate immunity in trophoblast functionality.
Clinical clamp assays and in vitro studies of placental perfusion demonstrate that TNF-α is the most significant independent predictor of insulin sensitivity in GDM patients, indicating close crosstalk between the immune and endocrine axis [145]. This immune interaction was corroborated in clinical association studies of GDM/obesity and maternal circulating levels of TNF-α and IL-6; this positive association remained after adjustment for total adipose mass [136,146,147,148,149][136][146][147][148][149]. Currently, it is known that cytokines such as TNF-α and IL-6 favor insulin resistance through inhibition of the insulin signaling cascade. Activation of the c-Jun N-terminal kinase (JNK) and IκB kinase (IKK) pathway targeted by TNF-α, increases serine phosphorylation of the IRS-1 and blocks normal tyrosine kinase activity of the IR [150]. On the other hand, IL-6 can activate the mammalian target of rapamycin (mTOR) which phosphorylates IRS-1 in serine residues, in a similar manner as TNF-α. Another molecular signaling of insulin resistance mediated by IL-6 is related to STAT-3 activation and the consequent activation of the Suppressor of Cytokine signaling (SOCS), which inhibits tyrosine phosphorylation of IRS-1. Both serine phosphorylation and blocked tyrosine phosphorylation of IRS-1 by inflammatory mediators impaired insulin action and subsequent insulin-regulated glucose uptake [151].
IL-1β is another important inflammatory mediator that has been shown to be increased in the placentae of obese mothers and with GDM [119,152,153][119][152][153]. In a first-trimester trophoblast cell line (Sw.71), high glucose levels (25 mM) increased trophoblast production of uric acid, which activated the inflammasome pathway, and positively regulated IL-1β release [154]. In mouse models for GDM, an increase in uterine and placental IL-1β levels and impaired glucose tolerance has been observed. In contrast, treatment with an anti-IL-1β antibody improved glucose tolerance in GDM mice [155]. Results obtained from in vitro models of hyperglycemia support the clinical data of the inflammatory environment in the placenta of women with GDM and provide evidence of the consequences that this generates on the anatomy and functionality of the placenta. Exposure of placental trophoblast cells to high concentrations of glucose (25 and 50 mM) significantly induced the secretion of cytokines and chemokines such as TNF-α, IL-1β, IL-6, IL-8, GRO-α, RANTES, and G-CSF [154]. Furthermore, high glucose concentrations suppress trophoblast viability and proliferation in vitro. These effects were mediated by the increase in miR-137, which in turn decreases the expression of the protein kinase activated by AMP (PRKAA1), positively regulating the placental secretion of IL-6 [156], and upregulating Bax/Bcl-2, COX-2, and caspase 9 expression [157].
It is well known that the migration of extravillous trophoblast into the maternal decidua and their interaction with endothelial cells from uterine spiral arteries are critical processes in placental development. In this sense, in vitro studies support an active intercommunication between trophoblasts and endothelial cells. The first-trimester trophoblast exposed to hyperglycemia (25 mM) induces the release of exosomes which in turn induces the endothelial release of IL-4, IL-6, IL-8, INF-γ, and TNF-α [158]. Together, the placental alteration of matrix metalloproteases, pro-angiogenic, and anti-angiogenic factors, as well as the shift towards a pro-inflammatory environment can explain the peripheral endothelial dysfunction in GDM [139,159,160][139][159][160]. Also, this endothelial imbalance may help to explain the observed high vasculogenesis rate, and capillary immaturity in GDM placentae [20[20][21],21], as will be discussed later in this review.
3.3. Altered Production of Adipokines by the GDM Placenta
Adipokines are bioactive polypeptides whose dysfunction is involved in inflammation, obesity, insulin resistance, and cardiovascular diseases [161]. Although adipokines have been described as secretion products of adipose tissue, the placenta can synthesize adipokines such as chemerin, omentin-1, visfatin, leptin, and adiponectin-like adipose tissue [8,135][8][135].
Leptin acts as a regulator of satiety and energy expenditure in the central nervous system [162]. Maternal plasma leptin levels increase in the first and second trimester of pregnancy and return to pre-pregnancy levels after delivery [163]. The placental synthesis of leptin is regulated by the different placental hormones such as β-hCG and estradiol [164,165][164][165]. Several studies have shown that the secretion of leptin by the placenta exerts autocrine actions stimulating the proliferation and survival of trophoblast cells [166]. Also, leptin increases placental lipid catabolism and vasodilation, possibly increasing the availability and transport of nutrients thus favoring fetal growth [167].
Regarding GDM, most researchers reported an increase in plasma and placental leptin levels [115,168,169,170][115][168][169][170] while other researchers did not observe a difference compared to healthy pregnancies [171]. In vitro evidence has shown that insulin induces leptin expression in trophoblast cells [162]. It has been found that maternal hyperglycemia in GDM regulates the leptin levels in umbilical cord blood generating macrosomia in the baby and increasing their risk of obesity in the future [168].
An increase of leptin and leptin receptor expression was found in the GDM placentae [172], contributing to the increased placental weight gain observed in GDM, along with the IGF-I axis as described before (see Figure 1). The binding of leptin with its receptor activates the signaling pathways MAPK, PI3K, and JaK-STAT which are also shared by IR [54]. In GDM placentae, the basal phosphorylation of STAT-3, MAPK 1/3, and Akt are increased, causing resistance to subsequent stimulation with Leptin or Insulin in vitro, suggesting crosstalk between insulin and leptin signaling in the human placenta [54].
In explants of the placenta, leptin significantly increases the release of IL-1β, IL-6, TNF-α, and prostaglandin E2 (PGE2) [173]. Similarly, leptin stimulates IL-6 secretion in trophoblast cells [174,175][174][175]. This increase in the production of pro-inflammatory cytokines evokes a chronic inflammatory milieu that in turn improves leptin release in the placenta, generating a vicious inflammatory loop [176].
Adiponectin is the second most studied adipokine. This peptide improves the IR signaling, makes lipid oxidation more efficient, inhibits the gluconeogenesis and the TNF-α signal in adipose tissue [177]. Adiponectin circulating levels increase during the first and second trimesters of normal pregnancy and later decrease post-partum [178]. Unlike leptin, low levels of adiponectin were found in maternal GDM serum compared to women with normal glucose tolerance [179,180][179][180]. It has been proposed that this decrease in serum adiponectin levels may serve as an early predictive factor for GDM development.
In terms of human placentae from pregnancies complicated with GDM, a significant downregulation of adiponectin mRNA and an upregulation of adiponectin receptor 1 (ADIPOR1) has been reported [181]. In the same study, it was observed that the cytokines INF-γ, TNF-α, and IL-6 differentially regulate the expression of adiponectin receptors (ADIPOR1 and ADIPOR2), as well as the expression and secretion of adiponectin. The researchers observed that placental adiponectin suppressed MAPK phosphorylation, particularly ERK1/2 and p38 that are essential for the onset of trophoblast differentiation, implantation, and placentation [181]. Furthermore, there is in vitro evidence that adiponectin promotes the trophoblast invasion by augmenting Matrix metalloproteinase (MMP)-2 and -9 production, and downregulating TIMP-2 mRNA expression [182]. Whereas adiponectin increases insulin sensitivity and modulates the invasion of trophoblasts, this adipokine could limit fetal and placental growth. Recent studies suggest that hypomethylation of the adiponectin gene in the placenta correlates with maternal insulin resistance and hyperglycemia, and with fetal macrosomia [183,184][183][184]. Secretion and expression of other placental adipokines have been described in women with GDM and obesity. Some studies showed an increase in many of these adipokines while others found no changes compared to women with normoglycemia [177]. More studies are needed to understand the impact of these adipokines on the development of GDM in the human.
3.4. Placental Innate Defense-Peptides in GDM
Regarding the innate immune system of the MPFU, the placenta, trophoblasts cells, decidual cells, stromal cells, and fetal membranes can produce several antimicrobial peptides, including human β-defensin (HBD)-1, HBD-2, HBD-3 and HBD-4, S100 proteins, human cathelicidin (hCTD) and human neutrophil peptides 1–4 (HNP 1–4) [185,186,187,188,189,190,191][185][186][187][188][189][190][191]. Moreover, the placenta also produces histones capable to neutralize certain bacterial endotoxins [192]. The main function of these defense peptides is to rapidly kill invader microorganisms; however, these multifunctional, amphipathic molecules also participate in angiogenesis, cell migration, and immune system modulation [193].
At this time, there is scarce information about the effect of diabetes and/or glucose levels on human placental antimicrobial peptides. In human amniotic epithelial cells, the high glucose culture medium is known to downregulate HBD2 production [194]. In other cell types such as macrophages, high glucose levels inhibit hCTD expression. However, in Mycobacterium tuberculosis-infected macrophages, hCTD levels increased as mycobacterial burden augmented, irrespective of the hyperglycemic environment [195]. Similarly, in a rat model of diabetes, lower levels of defensin BD1 have been found, but interestingly, they could be restored by insulin. On the contrary, defensin BD2 levels were found significantly higher than in non-diabetic animals, which was interpreted as the result of a glucose-dependent increased inflammatory state [196]. Likewise, in biopsies from diabetic foot ulcers, all studied defensins (HBD1-4) were overexpressed while hCTD was decreased in comparison to healthy skin. Remarkably, when an infectious agent was present, a significantly lower hCTD expression was observed. The authors concluded that the amount of antimicrobial peptide present in these diabetic tissues was not able to efficiently contain the infection [197].
Altogether, these results suggest that high blood glucose differentially regulates the expression of innate immune system components and that there seems to be an interaction between this hyperglycemic state and other assaults that may occur in diabetes, namely inflammation and infection. A particularly interesting case is that of hCTD, which is of primordial importance, especially for intracellular infections. In the human placenta, and similarly as in the cases just described, there is a differential inflammation-dependent regulation of hCTD and HBDs. Indeed, bacterial LPS endotoxin stimulated HBD2 and S100A9 mRNA levels, while significantly repressed basal and calcitriol-dependent hCTD expression [198].
References
- ACOG Practice Bulletin No. 190: Gestational Diabetes Mellitus. Obstet. Gynecol. 2018, 131, e49–e64.
- Hyperglycemia and Adverse Pregnancy Outcomes. N. Engl. J. Med. 2008, 358.
- Murray, S.R.; Reynolds, R.M. Short- and Long-Term Outcomes of Gestational Diabetes and Its Treatment on Fetal Development. Prenat. Diagn. 2020, 40.
- Bianco, M.E.; Josefson, J.L. Hyperglycemia During Pregnancy and Long-Term Offspring Outcomes. Curr. Diabetes Rep. 2019, 19.
- Dickens, L.T.; Thomas, C.C. Updates in Gestational Diabetes Prevalence, Treatment, and Health Policy. Curr. Diabetes Rep. 2019, 19.
- Brawerman, G.M.; Dolinsky, V.W. Therapies for Gestational Diabetes and Their Implications for Maternal and Offspring Health: Evidence from Human and Animal Studies. Pharmacol. Res. 2018, 130.
- McCance, D.R. Diabetes in Pregnancy: Management from Preconception to the Postnatal Period; NICE Guideline; NICE: Hoboken, NJ, USA, 2015; Volume 29, Available online: https://www.nice.org.uk/guidance/ng3/resources/diabetes-in-pregnancy-management-from-preconception-to-the-postnatal-period-pdf-51038446021 (accessed on 1 June 2021).
- Plows, J.F.; Stanley, J.L.; Baker, P.N.; Reynolds, C.M.; Vickers, M.H. The Pathophysiology of Gestational Diabetes Mellitus. Int. J. Mol. Sci. 2018, 19, 3342.
- Moyce, B.L.; Dolinsky, V.W. Maternal β-Cell Adaptations in Pregnancy and Placental Signalling: Implications for Gestational Diabetes. Int. J. Mol. Sci. 2018, 19, 3467.
- Dennison, R.A.; Chen, E.S.; Green, M.E.; Legard, C.; Kotecha, D.; Farmer, G.; Sharp, S.J.; Ward, R.J.; Usher-Smith, J.A.; Griffin, S.J. The Absolute and Relative Risk of Type 2 Diabetes after Gestational Diabetes: A Systematic Review and Meta-Analysis of 129 Studies. Diabetes Res. Clin. Pract. 2021, 171.
- American Diabetes Association. Management of Diabetes in Pregnancy: Standards of Medical Care in Diabetes—2020. Diabetes Care 2020, 43.
- Schäfer-Graf, U.M.; Gembruch, U.; Kainer, F.; Groten, T.; Hummel, S.; Hösli, I.; Grieshop, M.; Kaltheuner, M.; Bührer, C.; Kautzky-Willer, A.; et al. Gestational Diabetes Mellitus (GDM)—Diagnosis, Treatment and Follow-UpGuideline of the DDG and DGGG (S3 Level, AWMF Registry Number 057/008, February 2018). Geburtshilfe Frauenheilkd. 2018, 78.
- Alqudah, A.; McKinley, M.C.; McNally, R.; Graham, U.; Watson, C.J.; Lyons, T.J.; McClements, L. Risk of Pre-Eclampsia in Women Taking Metformin: A Systematic Review and Meta-Analysis. Diabet. Med. 2018, 35.
- Martis, R.; Crowther, C.A.; Shepherd, E.; Alsweiler, J.; Downie, M.R.; Brown, J. Treatments for Women with Gestational Diabetes Mellitus: An Overview of Cochrane Systematic Reviews. Cochrane Database Syst. Rev. 2018, 2018.
- Feng, Y.; Yang, H. Metformin–a Potentially Effective Drug for Gestational Diabetes Mellitus: A Systematic Review and Meta-Analysis. J. Matern. Fetal Neonatal Med. 2017, 30.
- Wang, X.; Liu, W.; Chen, H.; Chen, Q. Comparison of Insulin, Metformin, and Glyburide on Perinatal Complications of Gestational Diabetes Mellitus: A Systematic Review and Meta-Analysis. Gynecol. Obstet. Investig. 2021.
- Gui, J.; Liu, Q.; Feng, L. Metformin vs Insulin in the Management of Gestational Diabetes: A Meta-Analysis. PLoS ONE 2013, 8, e64585.
- Landi, S.N.; Radke, S.; Engel, S.M.; Boggess, K.; Stürmer, T.; Howe, A.S.; Funk, M.J. Association of Long-Term Child Growth and Developmental Outcomes with Metformin vs Insulin Treatment for Gestational Diabetes. JAMA Pediatr. 2019, 173.
- Wouldes, T.A.; Battin, M.; Coat, S.; Rush, E.C.; Hague, W.M.; Rowan, J.A. Neurodevelopmental Outcome at 2 Years in Offspring of Women Randomised to Metformin or Insulin Treatment for Gestational Diabetes. Arch. Dis. Child. Fetal Neonatal Ed. 2016, 101.
- Carrasco-Wong, I.; Moller, A.; Giachini, F.R.; Lima, V.V.; Toledo, F.; Stojanova, J.; Sobrevia, L.; San Martín, S. Placental Structure in Gestational Diabetes Mellitus. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866.
- Ehlers, E.; Talton, O.O.; Schust, D.J.; Schulz, L.C. Placental Structural Abnormalities in Gestational Diabetes and When They Develop: A Scoping Review. Placenta 2021.
- Lucas, M.J. Diabetes Complicating Pregnancy. Obstet. Gynecol. Clin. N. Am. 2001, 28.
- Huynh, J.; Dawson, D.; Roberts, D.; Bentley-Lewis, R. A Systematic Review of Placental Pathology in Maternal Diabetes Mellitus. Placenta 2015, 36.
- Alqudah, A.; Eastwood, K.A.; Jerotic, D.; Todd, N.; Hoch, D.; McNally, R.; Obradovic, D.; Dugalic, S.; Hunter, A.J.; Holmes, V.A.; et al. FKBPL and SIRT-1 Are Downregulated by Diabetes in Pregnancy Impacting on Angiogenesis and Endothelial Function. Front. Endocrinol. 2021, 12.
- Daskalakis, G.; Marinopoulos, S.; Krielesi, V.; Papapanagiotou, A.; Papantoniou, N.; Mesogitis, S.; Antsaklis, A. Placental Pathology in Women with Gestational Diabetes. Acta Obstet. Gynecol. Scand. 2008, 87.
- Madazli, R.; Tuten, A.; Calay, Z.; Uzun, H.; Uludag, S.; Ocak, V. The Incidence of Placental Abnormalities, Maternal and Cord Plasma Malondialdehyde and Vascular Endothelial Growth Factor Levels in Women with Gestational Diabetes Mellitus and Nondiabetic Controls. Gynecol. Obstet. Investig. 2008, 65.
- Fisher, J.J.; Vanderpeet, C.L.; Bartho, L.A.; McKeating, D.R.; Cuffe, J.S.M.; Holland, O.J.; Perkins, A.V. Mitochondrial Dysfunction in Placental Trophoblast Cells Experiencing Gestational Diabetes Mellitus. J. Physiol. 2021, 599.
- Valent, A.M.; Choi, H.; Kolahi, K.S.; Thornburg, K.L. Hyperglycemia and Gestational Diabetes Suppress Placental Glycolysis and Mitochondrial Function and Alter Lipid Processing. FASEB J. 2021, 35.
- Mishra, J.S.; Zhao, H.; Hattis, S.; Kumar, S. Elevated Glucose and Insulin Levels Decrease DHA Transfer across Human Trophoblasts via SIRT1-Dependent Mechanism. Nutrients 2020, 12, 1271.
- Hansis, C.; Grifo, J.A.; Tang, Y.X.; Krey, L.C. Assessment of Beta-HCG, Beta-LH MRNA and Ploidy in Individual Human Blastomeres. Reprod. Biomed. Online 2002, 5.
- Butler, S.A.; Luttoo, J.; Freire, M.O.T.; Abban, T.K.; Borrelli, P.T.A.; Iles, R.K. Human Chorionic Gonadotropin (HCG) in the Secretome of Cultured Embryos: Hyperglycosylated HCG and HCG-Free Beta Subunit Are Potential Markers for Infertility Management and Treatment. Reprod. Sci. 2013, 20.
- Feldt-Rasmussen, U.; Mathiesen, E.R. Endocrine Disorders in Pregnancy: Physiological and Hormonal Aspects of Pregnancy. Best Pract. Res. Clin. Endocrinol. Metab. 2011, 25.
- Rout, M.; Lulu, S.S. Molecular and Disease Association of Gestational Diabetes Mellitus Affected Mother and Placental Datasets Reveal a Strong Link between Insulin Growth Factor (IGF) Genes in Amino Acid Transport Pathway: A Network Biology Approach. J. Cell. Biochem. 2019, 120.
- Alur, V.; Raju, V.; Vastrad, B.; Tengli, A.; Vastrad, C.; Kotturshetti, S. Integrated Bioinformatics Analysis Reveals Novel Key Biomarkers and Potential Candidate Small Molecule Drugs in Gestational Diabetes Mellitus. Biosci. Rep. 2021, 41.
- Yang, Y.; Guo, F.; Peng, Y.; Chen, R.; Zhou, W.; Wang, H.; OuYang, J.; Yu, B.; Xu, Z. Transcriptomic Profiling of Human Placenta in Gestational Diabetes Mellitus at the Single-Cell Level. Front. Endocrinol. 2021, 12.
- Westermeier, F.; Sáez, T.; Arroyo, P.; Toledo, F.; Gutiérrez, J.; Sanhueza, C.; Pardo, F.; Leiva, A.; Sobrevia, L. Insulin Receptor Isoforms: An Integrated View Focused on Gestational Diabetes Mellitus. Diabetes Metab. Res. Rev. 2016, 32.
- Sobrevia, L.; Salsoso, R.; Fuenzalida, B.; Barros, E.; Toledo, L.; Silva, L.; Pizarro, C.; Subiabre, M.; Villalobos, R.; Araos, J.; et al. Insulin Is a Key Modulator of Fetoplacental Endothelium Metabolic Disturbances in Gestational Diabetes Mellitus. Front. Physiol. 2016, 7.
- Larner, J.; Huang, L.C.; Tang, G.; Suzuki, S.; Schwartz, C.F.W.; Romero, G.; Roulidis, Z.; Zeller, K.; Shen, T.Y.; Oswald, A.S.; et al. Insulin Mediators: Structure and Formation. Cold Spring Harb. Symp. Quant. Biol. 1988, 53.
- Cross, D.A.E.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of Glycogen Synthase Kinase-3 by Insulin Mediated by Protein Kinase B. Nature 1995, 378.
- Kohn, A.D.; Kovacina, K.S.; Roth, R.A. Insulin Stimulates the Kinase Activity of RAC-PK, a Pleckstrin Homology Domain Containing Ser/Thr Kinase. EMBO J. 1995, 14.
- Batista, T.M.; Haider, N.; Kahn, C.R. Defining the Underlying Defect in Insulin Action in Type 2 Diabetes. Diabetologia 2021, 64.
- O’Neill, B.T.; Lee, K.Y.; Klaus, K.; Softic, S.; Krumpoch, M.T.; Fentz, J.; Stanford, K.I.; Robinson, M.M.; Cai, W.; Kleinridders, A.; et al. Insulin and IGF-1 Receptors Regulate FoxO-Mediated Signaling in Muscle Proteostasis. J. Clin. Investig. 2016, 126.
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 Is an Insulin-Regulated Inhibitor of the MTORC1 Protein Kinase. Mol. Cell 2007, 25.
- Tee, A.R.; Fingar, D.C.; Manning, B.D.; Kwiatkowski, D.J.; Cantley, L.C.; Blenis, J. Tuberous Sclerosis Complex-1 and -2 Gene Products Function Together to Inhibit Mammalian Target of Rapamycin (MTOR)-Mediated Downstream Signaling. Proc. Natl. Acad. Sci. USA 2002, 99.
- Sano, H.; Kane, S.; Sano, E.; Mîinea, C.P.; Asara, J.M.; Lane, W.S.; Garner, C.W.; Lienhard, G.E. Insulin-Stimulated Phosphorylation of a Rab GTPase-Activating Protein Regulates GLUT4 Translocation. J. Biol. Chem. 2003, 278.
- Avruch, J.; Khokhlatchev, A.; Kyriakis, J.M.; Luo, Z.; Tzivion, G.; Vavvas, D.; Zhang, X.F. Ras Activation of the Raf Kinase: Tyrosine Kinase Recruitment of the MAP Kinase Cascade. Recent Prog. Horm. Res. 2001, 56.
- Sesti, G.; Tullio, A.N.; D’Alfonso, R.; Napolitano, M.L.; Marini, M.A.; Borboni, P.; Longhi, R.; Albonici, L.; Fusco, A.; Aglianò, A.M.; et al. Tissue-Specific Expression of Two Alternatively Spliced Isoforms of the Human Insulin Receptor Protein. Acta Diabetol. 1994, 31.
- Illsley, N.P.; Baumann, M.U. Human Placental Glucose Transport in Fetoplacental Growth and Metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866.
- Stanirowski, P.J.; Szukiewicz, D.; Pazura-Turowska, M.; Sawicki, W.; Cendrowski, K. Placental Expression of Glucose Transporter Proteins in Pregnancies Complicated by Gestational and Pregestational Diabetes Mellitus. Can. J. Diabetes 2018, 42.
- Hiden, U.; Maier, A.; Bilban, M.; Ghaffari-Tabrizi, N.; Wadsack, C.; Lang, I.; Dohr, G.; Desoye, G. Insulin Control of Placental Gene Expression Shifts from Mother to Foetus over the Course of Pregnancy. Diabetologia 2006, 49.
- Alonso, A.; del Rey, C.G.; Navarro, A.; Tolivia, J.; González, C.G. Effects of Gestational Diabetes Mellitus on Proteins Implicated in Insulin Signaling in Human Placenta. Gynecol. Endocrinol. 2006, 22.
- Balachandiran, M.; Bobby, Z.; Dorairajan, G.; Gladwin, V.; Vinayagam, V.; Packirisamy, R.M. Decreased Maternal Serum Adiponectin and Increased Insulin-like Growth Factor-1 Levels along with Increased Placental Glucose Transporter-1 Expression in Gestational Diabetes Mellitus: Possible Role in Fetal Overgrowth: Regulation of Placental GLUT-1 Expression in Gestational Diabetes Mellitus. Placenta 2021, 104.
- Colomiere, M.; Permezel, M.; Riley, C.; Desoye, G.; Lappas, M. Defective Insulin Signaling in Placenta from Pregnancies Complicated by Gestational Diabetes Mellitus. Eur. J. Endocrinol. 2009, 160.
- Pérez-Pérez, A.; Guadix, P.; Maymó, J.; Dueñas, J.L.; Varone, C.; Fernández-Sánchez, M.; Sánchez-Margalet, V. Insulin and Leptin Signaling in Placenta from Gestational Diabetic Subjects. Horm. Metab. Res. 2015, 48.
- Gęca, T.; Kwaśniewska, A. The Influence of Gestational Diabetes Mellitus upon the Selected Parameters of the Maternal and Fetal System of Insulin-Like Growth Factors (IGF-1, IGF-2, IGFBP1-3)—A Review and a Clinical Study. J. Clin. Med. 2020, 9, 3256.
- Osmond, D.T.D.; King, R.G.; Brennecke, S.P.; Gude, N.M. Placental Glucose Transport and Utilisation Is Altered at Term in Insulin-Treated, Gestational-Diabetic Patients. Diabetologia 2001, 44.
- Osmond, D.T.D.; Nolan, C.J.; King, R.G.; Brennecke, S.P.; Gude, N.M. Effects of Gestational Diabetes on Human Placental Glucose Uptake, Transfer, and Utilisation. Diabetologia 2000, 43.
- Tumminia, A.; Scalisi, N.M.; Milluzzo, A.; Ettore, G.; Vigneri, R.; Sciacca, L. Maternal Diabetes Impairs Insulin and IGF-1 Receptor Expression and Signaling in Human Placenta. Front. Endocrinol. 2021, 12.
- Forbes, B.E.; Blyth, A.J.; Wit, J.M. Disorders of IGFs and IGF-1R Signaling Pathways. Mol. Cell. Endocrinol. 2020, 518.
- Wang, X.R.; Wang, W.J.; Yu, X.; Hua, X.; Ouyang, F.; Luo, Z.C. Insulin-like Growth Factor Axis Biomarkers and Gestational Diabetes Mellitus: A Systematic Review and Meta-Analysis. Front. Endocrinol. 2019, 10.
- Frasca, F.; Pandini, G.; Scalia, P.; Sciacca, L.; Mineo, R.; Costantino, A.; Goldfine, I.D.; Belfiore, A.; Vigneri, R. Insulin Receptor Isoform A, a Newly Recognized, High-Affinity Insulin-Like Growth Factor II Receptor in Fetal and Cancer Cells. Mol. Cell. Biol. 1999, 19.
- Blanquart, C.; Achi, J.; Issad, T. Characterization of IRA/IRB Hybrid Insulin Receptors Using Bioluminescence Resonance Energy Transfer. Biochem. Pharmacol. 2008, 76.
- Federici, M.; Porzio, O.; Zucaro, L.; Fusco, A.; Borboni, P.; Lauro, D.; Sesti, G. Distribution of Insulin/Insulin-like Growth Factor-I Hybrid Receptors in Human Tissues. Mol. Cell. Endocrinol. 1997, 129.
- Benyoucef, S.; Surinya, K.H.; Hadaschik, D.; Siddle, K. Characterization of Insulin/IGF Hybrid Receptors: Contributions of the Insulin Receptor L2 and Fn1 Domains and the Alternatively Spliced Exon 11 Sequence to Ligand Binding and Receptor Activation. Biochem. J. 2007, 403.
- Blyth, A.J.; Kirk, N.S.; Forbes, B.E. Understanding IGF-II Action through Insights into Receptor Binding and Activation. Cells 2020, 9, 2276.
- Han, V.K.; Bassett, N.; Walton, J.; Challis, J.R. The Expression of Insulin-like Growth Factor (IGF) and IGF-Binding Protein (IGFBP) Genes in the Human Placenta and Membranes: Evidence for IGF-IGFBP Interactions at the Feto-Maternal Interface. J. Clin. Endocrinol. Metab. 1996, 81.
- Holt, R.I.G.; Simpson, H.L.; Sönksen, P.H. The Role of the Growth Hormone-Insulin-like Growth Factor Axis in Glucose Homeostasis. Diabet. Med. 2003, 20.
- Baumann, M.U.; Schneider, H.; Malek, A.; Palta, V.; Surbek, D.V.; Sager, R.; Zamudio, S.; Illsley, N.P. Regulation of Human Trophoblast GLUT1 Glucose Transporter by Insulin-Like Growth Factor I (IGF-I). PLoS ONE 2014, 9, e106037.
- Scavo, L.M.; Karas, M.; Murray, M.; Leroith, D. Insulin-like Growth Factor-I Stimulates Both Cell Growth and Lipogenesis during Differentiation of Human Mesenchymal Stem Cells into Adipocytes. J. Clin. Endocrinol. Metab. 2004, 89.
- Clemmons, D.R.; Sleevi, M.; Allan, G.; Sommer, A. Effects of Combined Recombinant Insulin-like Growth Factor (IGF)-I and IGF Binding Protein-3 in Type 2 Diabetic Patients on Glycemic Control and Distribution of IGF-I and IGF-II among Serum Binding Protein Complexes. J. Clin. Endocrinol. Metab. 2007, 92.
- Devedjian, J.C.; George, M.; Casellas, A.; Pujol, A.; Visa, J.; Pelegrín, M.; Gros, L.; Bosch, F. Transgenic Mice Overexpressing Insulin-like Growth Factor-II in β Cells Develop Type 2 Diabetes. J. Clin. Investig. 2000, 105.
- Dabelea, D.; Crume, T. Maternal Environment and the Transgenerational Cycle of Obesity and Diabetes. Diabetes 2011, 60.
- Stanirowski, P.J.; Szukiewicz, D.; Pyzlak, M.; Abdalla, N.; Sawicki, W.; Cendrowski, K. Impact of Pre-Gestational and Gestational Diabetes Mellitus on the Expression of Glucose Transporters GLUT-1, GLUT-4 and GLUT-9 in Human Term Placenta. Endocrine 2017, 55.
- Gaither, K.; Quraishi, A.N.; Illsley, N.P. Diabetes Alters the Expression and Activity of the Human Placental GLUT1 Glucose Transporter 1. J. Clin. Endocrinol. Metab. 1999, 84.
- Borges, M.H.; Pullockaran, J.; Catalano, P.M.; Baumann, M.U.; Zamudio, S.; Illsley, N.P. Human Placental GLUT1 Glucose Transporter Expression and the Fetal Insulin-like Growth Factor Axis in Pregnancies Complicated by Diabetes. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865.
- Lappas, M. Insulin-like Growth Factor-Binding Protein 1 and 7 Concentrations Are Lower in Obese Pregnant Women, Women with Gestational Diabetes and Their Fetuses. J. Perinatol. 2015, 35.
- Bach, L.A. Current Ideas on the Biology of IGFBP-6: More than an IGF-II Inhibitor? Growth Horm. IGF Res. 2016, 30–31.
- Shang, M.; Wen, Z. Increased Placental IGF-1/MTOR Activity in Macrosomia Born to Women with Gestational Diabetes. Diabetes Res. Clin. Pract. 2018, 146.
- Liu, K.; Wu, H.Y.; Xu, Y.H. Study on the Relationship between the Expression of IGF-1 in Umbilical Cord Blood and Abnormal Glucose Metabolism during Pregnancy. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 647–651.
- Federici, M.; Zucaro, L.; Porzio, O.; Massoud, R.; Borboni, P.; Lauro, D.; Sesti, G. Increased Expression of Insulin/Insulin-like Growth Factor-1 Hybrid Receptors in Skeletal Muscle of Noninsulin-Dependent Diabetes Mellitus Subjects. J. Clin. Investig. 1996, 98.
- Federici, M.; Porzio, O.; Zucaro, L.; Giovannone, B.; Borboni, P.; Marini, M.A.; Lauro, D.; Sesti, G. Increased Abundance of Insulin/IGF-I Hybrid Receptors in Adipose Tissue from NIDDM Patients. Mol. Cell. Endocrinol. 1997, 135.
- Valensise, H.; Liu, Y.Y.; Federici, M.; Lauro, D.; Dell’anna, D.; Romanini, C.; Sesti, G. Increased Expression of Low-Affinity Insulin Receptor Isoform and Insulin/Insulin-like Growth Factor-1 Hybrid Receptors in Term Placenta from Insulin-Resistant Women with Gestational Hypertension. Diabetologia 1996, 39.
- Pandini, G.; Vigneri, R.; Costantino, A.; Frasca, F.; Ippolito, A.; Fujita-Yamaguchi, Y.; Siddle, K.; Goldfine, I.D.; Belfiore, A. Insulin and Insulin-like Growth Factor-I (IGF-I) Receptor Overexpression in Breast Cancers Leads to Insulin/IGF-I Hybrid Receptor Overexpression: Evidence for a Second Mechanism of IGF-I Signaling. Clin. Cancer Res. 1999, 5, 1935–1944.
- Catalano, P.M.; Tyzbir, E.D.; Roman, N.M.; Amini, S.B.; Sims, E.A.H. Longitudinal Changes in Insulin Release and Insulin Resistance in Nonobese Pregnant Women. Am. J. Obstet. Gynecol. 1991, 165.
- Stefanoska, I.; Jovanović Krivokuća, M.; Vasilijić, S.; Ćujić, D.; Vićovac, L. Prolactin Stimulates Cell Migration and Invasion by Human Trophoblast in Vitro. Placenta 2013, 34.
- Binart, N. Prolactin and Pregnancy in Mice and Humans. Ann. Endocrinol. 2016, 77.
- Garzia, E.; Clauser, R.; Persani, L.; Borgato, S.; Bulfamante, G.; Avagliano, L.; Quadrelli, F.; Marconi, A.M. Prolactin and Proinflammatory Cytokine Expression at the Fetomaternal Interface in First Trimester Miscarriage. Fertil. Steril. 2013, 100.
- Cattini, P.A.; Jin, Y.; Jarmasz, J.S.; Noorjahan, N.; Bock, M.E. Obesity and Regulation of Human Placental Lactogen Production in Pregnancy. J. Neuroendocrinol. 2020, 32, e12859.
- Newbern, D.; Freemark, M. Placental Hormones and the Control of Maternal Metabolism and Fetal Growth. Curr. Opin. Endocrinol. Diabetes Obes. 2011, 18.
- Hu, Z.-Z.; Zhuang, L.; Meng, J.; Tsai-Morris, C.-H.; Dufau, M.L. Complex 5′ Genomic Structure of the Human Prolactin Receptor: Multiple Alternative Exons 1 and Promoter Utilization. Endocrinology 2002, 143.
- Abramicheva, P.A.; Smirnova, O.V. Prolactin Receptor Isoforms as the Basis of Tissue-Specific Action of Prolactin in the Norm and Pathology. Biochemistry 2019, 84.
- Sorenson, R.L.; Brelje, T.C. Prolactin Receptors Are Critical to the Adaptation of Islets to Pregnancy. Endocrinology 2009, 150.
- Yan, L.; Figueroa, D.J.; Austin, C.P.; Liu, Y.; Bugianesi, R.M.; Slaughter, R.S.; Kaczorowski, G.J.; Kohler, M.G. Expression of Voltage-Gated Potassium Channels in Human and Rhesus Pancreatic Islets. Diabetes 2004, 53.
- Huang, C.; Snider, F.; Cross, J.C. Prolactin Receptor Is Required for Normal Glucose Homeostasis and Modulation of β-Cell Mass during Pregnancy. Endocrinology 2009, 150.
- Kim, H.; Toyofuku, Y.; Lynn, F.C.; Chak, E.; Uchida, T.; Mizukami, H.; Fujitani, Y.; Kawamori, R.; Miyatsuka, T.; Kosaka, Y.; et al. Serotonin Regulates Pancreatic Beta Cell Mass during Pregnancy. Nat. Med. 2010, 16.
- Ben-Jonathan, N.; LaPensee, C.R.; LaPensee, E.W. What Can We Learn from Rodents about Prolactin in Humans? Endocr. Rev. 2008, 29.
- Brelje, T.C.; Scharp, D.W.; Lacy, P.E.; Ogren, L.; Talamantes, F.; Robertson, M.; Friesen, H.G.; Sorenson, R.L. Effect of Homologous Placental Lactogens, Prolactins, and Growth Hormones on Islet b-Cell Division and Insulin Secretion in Rat, Mouse, and Human Islets: Implication for Placental Lactogen Regulation of Islet Function during Pregnancy. Endocrinology 1993, 132.
- Ernst, S.; Demirci, C.; Valle, S.; Velazquez-Garcia, S.; Garcia-Ocaña, A. Mechanisms in the Adaptation of Maternal β-Cells during Pregnancy. Diabetes Manag. 2011, 1.
- Demirci, C.; Ernst, S.; Alvarez-Perez, J.C.; Rosa, T.; Valle, S.; Shridhar, V.; Casinelli, G.P.; Alonso, L.C.; Vasavada, R.C.; García-Ocana, A. Loss of HGF/c-Met Signaling in Pancreatic β-Cells Leads to Incomplete Maternal β-Cell Adaptation and Gestational Diabetes Mellitus. Diabetes 2012, 61.
- Hakonen, E.; Ustinov, J.; Palgi, J.; Miettinen, P.J.; Otonkoski, T. EGFR Signaling Promotes β-Cell Proliferation and Survivin Expression during Pregnancy. PLoS ONE 2014, 9, e93651.
- Sergeev, I.N.; Rhoten, W.B. 1, 25-Dihydroxyvitamin D3 Evokes Oscillations of Intracellular Calcium in a Pancreatic β-Cell Line. Endocrinology 1995, 136.
- Altuntaś, S.Ç.; Evran, M.; Sert, M.; Tetiker, T. Markers of Metabolic Syndrome in Patients with Pituitary Adenoma: A Case Series of 303 Patients. Horm. Metab. Res. 2019, 51.
- Le, T.N.; Elsea, S.H.; Romero, R.; Chaiworapongsa, T.; Francis, G.L. Prolactin Receptor Gene Polymorphisms Are Associated with Gestational Diabetes. Genet. Test. Mol. Biomark. 2013, 17.
- Ursell, W.; Brudenell, M.; Chard, T. Placental Lactogen Levels in Diabetic Pregnancy. Br. Med. J. 1973, 2, 80–82.
- Skouby, S.O.; Kühl, C.; Hornnes, P.J.; Andersen, A.N. Prolactin and Glucose Tolerance in Normal and Gestational Diabetic Pregnancy. Obstet. Gynecol. 1986, 67, 17–20.
- Macotela, Y.; Triebel, J.; Clapp, C. Time for a New Perspective on Prolactin in Metabolism. Trends Endocrinol. Metab. 2020, 31.
- Park, S.; Kim, D.S.; Daily, J.W.; Kim, S.H. Serum Prolactin Concentrations Determine Whether They Improve or Impair β-Cell Function and Insulin Sensitivity in Diabetic Rats. Diabetes Metab. Res. Rev. 2011, 27.
- Kawaharada, R.; Masuda, H.; Chen, Z.; Blough, E.; Kohama, T.; Nakamura, A. Intrauterine Hyperglycemia-Induced Inflammatory Signalling via the Receptor for Advanced Glycation End Products in the Cardiac Muscle of the Infants of Diabetic Mother Rats. Eur. J. Nutr. 2018, 57.
- Pantham, P.; Aye, I.L.M.H.; Powell, T.L. Inflammation in Maternal Obesity and Gestational Diabetes Mellitus. Placenta 2015, 36.
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory Mechanisms in Obesity. Annu. Rev. Immunol. 2011, 29.
- Yessoufou, A.; Moutairou, K. Maternal Diabetes in Pregnancy: Early and Long-Term Outcomes on the Offspring and the Concept of “Metabolic Memory”. Exp. Diabetes Res. 2011, 2011.
- Westermeier, F.; Sáez, P.J.; Villalobos-Labra, R.; Sobrevia, L.; Farías-Jofré, M. Programming of Fetal Insulin Resistance in Pregnancies with Maternal Obesity by ER Stress and Inflammation. BioMed Res. Int. 2014, 2014.
- Dudele, A.; Hougaard, K.S.; Kjølby, M.; Hokland, M.; Winther, G.; Elfving, B.; Wegener, G.; Nielsen, A.L.; Larsen, A.; Nøhr, M.K.; et al. Chronic Maternal Inflammation or High-Fat-Feeding Programs Offspring Obesity in a Sex-Dependent Manner. Int. J. Obes. 2017, 41.
- Perrin, E.M.; O’Shea, T.M.; Skinner, A.C.; Bose, C.; Allred, E.N.; Fichorova, R.N.; van der Burg, J.W.; Leviton, A. Elevations of Inflammatory Proteins in Neonatal Blood Are Associated with Obesity and Overweight among 2-Year-Old Children Born Extremely Premature. Pediatr. Res. 2018, 83.
- Tsiotra, P.C.; Halvatsiotis, P.; Patsouras, K.; Maratou, E.; Salamalekis, G.; Raptis, S.A.; Dimitriadis, G.; Boutati, E. Circulating Adipokines and MRNA Expression in Adipose Tissue and the Placenta in Women with Gestational Diabetes Mellitus. Peptides 2018, 101.
- Zhang, J.; Chi, H.; Xiao, H.; Tian, X.; Wang, Y.; Yun, X.; Xu, Y. Interleukin 6 (IL-6) and Tumor Necrosis Factor α (TNF-α) Single Nucleotide Polymorphisms (SNPs), Inflammation and Metabolism in Gestational Diabetes Mellitus in Inner Mongolia. Med. Sci. Monit. 2017, 23.
- Angelo, A.G.S.; Neves, C.T.C.; Lobo, T.F.; Godoy, R.V.C.; Ono, É.; Mattar, R.; Daher, S. Monocyte Profile in Peripheral Blood of Gestational Diabetes Mellitus Patients. Cytokine 2018, 107.
- Lekva, T.; Michelsen, A.E.; Aukrust, P.; Paasche Roland, M.C.; Henriksen, T.; Bollerslev, J.; Ueland, T. CXC Chemokine Ligand 16 Is Increased in Gestational Diabetes Mellitus and Preeclampsia and Associated with Lipoproteins in Gestational Diabetes Mellitus at 5 Years Follow-Up. Diabetes Vasc. Dis. Res. 2017, 14.
- Hara, C.D.C.P.; França, E.L.; Fagundes, D.L.G.; de Queiroz, A.A.; Rudge, M.V.C.; Honorio-França, A.C.; Calderon, I.D.M.P. Characterization of Natural Killer Cells and Cytokines in Maternal Placenta and Fetus of Diabetic Mothers. J. Immunol. Res. 2016, 2016.
- Mrizak, I.; Grissa, O.; Henault, B.; Fekih, M.; Bouslema, A.; Boumaiza, I.; Zaouali, M.; Tabka, Z.; Khan, N.A. Placental Infiltration of Inflammatory Markers in Gestational Diabetic Women. Gen. Physiol. Biophys. 2014, 33.
- Tchirikov, M.; Schlabritz-Loutsevitch, N.; Maher, J.; Buchmann, J.; Naberezhnev, Y.; Winarno, A.S.; Seliger, G. Mid-Trimester Preterm Premature Rupture of Membranes (PPROM): Etiology, Diagnosis, Classification, International Recommendations of Treatment Options and Outcome. J. Perinat. Med. 2018, 46.
- Toniolo, A.; Cassani, G.; Puggioni, A.; Rossi, A.; Colombo, A.; Onodera, T.; Ferrannini, E. The Diabetes Pandemic and Associated Infections: Suggestions for Clinical Microbiology. Rev. Med. Microbiol. 2019, 30.
- Yu, J.; Zhou, Y.; Gui, J.; Li, A.Z.; Su, X.L.; Feng, L. Assessment of the Number and Function of Macrophages in the Placenta of Gestational Diabetes Mellitus Patients. J. Huazhong Univ. Sci. Technol. Med. Sci. 2013, 33.
- Zheng, L.; Li, C.; Qi, W.; Qiao, B.; Zhao, H.; Zhou, Y.; Lu, C. Expression of Macrophage Migration Inhibitory Factor Gene in Placenta Tissue and Its Correlation with Gestational Diabetes Mellitus. Natl. Med. J. China 2017, 97.
- Sisino, G.; Bouckenooghe, T.; Aurientis, S.; Fontaine, P.; Storme, L.; Vambergue, A. Diabetes during Pregnancy Influences Hofbauer Cells, a Subtype of Placental Macrophages, to Acquire a pro-Inflammatory Phenotype. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832.
- Dasu, M.R.; Jialal, I. Free Fatty Acids in the Presence of High Glucose Amplify Monocyte Inflammation via Toll-like Receptors. Am. J. Physiol. Endocrinol. Metab. 2011, 300.
- Schliefsteiner, C.; Peinhaupt, M.; Kopp, S.; Lögl, J.; Lang-Olip, I.; Hiden, U.; Heinemann, A.; Desoye, G.; Wadsack, C. Human Placental Hofbauer Cells Maintain an Anti-Inflammatory M2 Phenotype despite the Presence of Gestational Diabetes Mellitus. Front. Immunol. 2017, 8.
- Stoikou, M.; Grimolizzi, F.; Giaglis, S.; Schäfer, G.; van Breda, S.V.; Hoesli, I.M.; Lapaire, O.; Huhn, E.A.; Hasler, P.; Rossi, S.W.; et al. Gestational Diabetes Mellitus Is Associated with Altered Neutrophil Activity. Front. Immunol. 2017, 8.
- Menegazzo, L.; Ciciliot, S.; Poncina, N.; Mazzucato, M.; Persano, M.; Bonora, B.; Albiero, M.; Vigili de Kreutzenberg, S.; Avogaro, A.; Fadini, G.P. NETosis Is Induced by High Glucose and Associated with Type 2 Diabetes. Acta Diabetol. 2015, 52.
- Sun, T.; Meng, F.; Zhao, H.; Yang, M.; Zhang, R.; Yu, Z.; Huang, X.; Ding, H.; Liu, J.; Zang, S. Elevated First-Trimester Neutrophil Count Is Closely Associated with the Development of Maternal Gestational Diabetes Mellitus and Adverse Pregnancy Outcomes. Diabetes 2020, 69.
- Vorobjeva, N.V.; Pinegin, B.V. Neutrophil Extracellular Traps: Mechanisms of Formation and Role in Health and Disease. Biochemistry 2014, 79.
- Olmos-Ortiz, A.; Flores-Espinosa, P.; Mancilla-Herrera, I.; Vega-Sánchez, R.; Díaz, L.; Zaga-Clavellina, V. Innate Immune Cells and Toll-like Receptor–Dependent Responses at the Maternal–Fetal Interface. Int. J. Mol. Sci. 2019, 20, 3654.
- Shmeleva, E.V.; Colucci, F. Maternal Natural Killer Cells at the Intersection between Reproduction and Mucosal Immunity. Mucosal Immunol. 2021.
- Lobo, T.F.; Borges, C.D.M.; Mattar, R.; Gomes, C.P.; de Angelo, A.G.S.; Pendeloski, K.P.T.; Daher, S. Impaired Treg and NK Cells Profile in Overweight Women with Gestational Diabetes Mellitus. Am. J. Reprod. Immunol. 2018, 79.
- Catalano, P.M.; Huston, L.; Amini, S.B.; Kalhan, S.C. Longitudinal Changes in Glucose Metabolism during Pregnancy in Obese Women with Normal Glucose Tolerance and Gestational Diabetes Mellitus. Am. J. Obstet. Gynecol. 1999, 180.
- Atègbo, J.M.; Grissa, O.; Yessoufou, A.; Hichami, A.; Dramane, K.L.; Moutairou, K.; Miled, A.; Grissa, A.; Jerbi, M.; Tabka, Z.; et al. Modulation of Adipokines and Cytokines in Gestational Diabetes and Macrosomia. J. Clin. Endocrinol. Metab. 2006, 91.
- Bari, M.F.; Weickert, M.O.; Sivakumar, K.; James, S.G.; Snead, D.R.J.; Tan, B.K.; Randeva, H.S.; Bastie, C.C.; Vatish, M. Elevated Soluble CD163 in Gestational Diabetes Mellitus: Secretion from Human Placenta and Adipose Tissue. PLoS ONE 2014, 9, e101327.
- Lacroix, M.; Lizotte, F.; Hivert, M.-F.; Geraldes, P.; Perron, P. Calcifediol Decreases Interleukin-6 Secretion by Cultured Human Trophoblasts from GDM Pregnancies. J. Endocr. Soc. 2019, 3.
- Radaelli, T.; Varastehpour, A.; Catalano, P.; Hauguel-De Mouzon, S. Gestational Diabetes Induces Placental Genes for Chronic Stress and Inflammatory Pathways. Diabetes 2003, 52.
- Li, J.; Yin, Q.; Wu, H. Structural basis of signal transduction in the TNF receptor superfamily. Adv. Immunol. 2013, 119.
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFκB System. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8.
- Lumeng, C.N. Innate Immune Activation in Obesity. Mol. Asp. Med. 2013, 34.
- Feng, H.; Su, R.; Song, Y.; Wang, C.; Lin, L.; Ma, J.; Yang, H. Positive Correlation between Enhanced Expression of TLR4/MyD88/NF-ΚB with Insulin Resistance in Placentae of Gestational Diabetes Mellitus. PLoS ONE 2016, 11, e0157185.
- Corrêa-Silva, S.; Alencar, A.P.; Moreli, J.B.; Borbely, A.U.; Lima, L.D.S.; Scavone, C.; Damasceno, D.C.; Rudge, M.V.C.; Bevilacqua, E.; Calderon, I.M.P. Hyperglycemia Induces Inflammatory Mediators in the Human Chorionic Villous. Cytokine 2018, 111.
- Kirwan, J.P.; Hauguel-De Mouzon, S.; Lepercq, J.; Challier, J.C.; Huston-Presley, L.; Friedman, J.E.; Kalhan, S.C.; Catalano, P.M. TNF-α Is a Predictor of Insulin Resistance in Human Pregnancy. Diabetes 2002, 51.
- Friis, C.M.; Roland, M.C.P.; Godang, K.; Ueland, T.; Tanbo, T.; Bollerslev, J.; Henriksen, T. Adiposity-Related Inflammation: Effects of Pregnancy. Obesity 2013, 21.
- Vega-Sanchez, R.; Barajas-Vega, H.A.; Rozada, G.; Espejel-Nuñez, A.; Beltran-Montoya, J.; Vadillo-Ortega, F. Association between Adiposity and Inflammatory Markers in Maternal and Fetal Blood in a Group of Mexican Pregnant Women. Br. J. Nutr. 2010, 104.
- Aye, I.L.M.H.; Lager, S.; Ramirez, V.I.; Gaccioli, F.; Dudley, D.J.; Jansson, T.; Powell, T.L. Increasing Maternal Body Mass Index Is Associated with Systemic Inflammation in the Mother and the Activation of Distinct Placental Inflammatory Pathways. Biol. Reprod. 2014, 90.
- Mohammed, A.; Aliyu, I.S. Maternal Serum Level of TNF-α in Nigerian Women with Gestational Diabetes Mellitus. Pan Afr. Med. J. 2018, 31.
- Catalano, P.M. Obesity, Insulin Resistance, and Pregnancy Outcome. Reproduction 2010, 140.
- Carey, A.L.; Febbraio, M.A. Interleukin-6 and Insulin Sensitivity: Friend or Foe? Diabetologia 2004, 47.
- Challier, J.C.; Basu, S.; Bintein, T.; Minium, J.; Hotmire, K.; Catalano, P.M.; Hauguel-de Mouzon, S. Obesity in Pregnancy Stimulates Macrophage Accumulation and Inflammation in the Placenta. Placenta 2008, 29.
- Stirm, L.; Kovářová, M.; Perschbacher, S.; Michlmaier, R.; Fritsche, L.; Siegel-Axel, D.; Schleicher, E.; Peter, A.; Pauluschke-Fröhlich, J.; Brucker, S.; et al. BMI-Independent Effects of Gestational Diabetes on Human Placenta. J. Clin. Endocrinol. Metab. 2018, 103.
- Han, C.S.; Herrin, M.A.; Pitruzzello, M.C.; Mulla, M.J.; Werner, E.F.; Pettker, C.M.; Flannery, C.A.; Abrahams, V.M. Glucose and Metformin Modulate Human First Trimester Trophoblast Function: A Model and Potential Therapy for Diabetes-Associated Uteroplacental Insufficiency. Am. J. Reprod. Immunol. 2015, 73.
- Schulze, F.; Wehner, J.; Kratschmar, D.V.; Makshana, V.; Meier, D.T.; Häuselmann, S.P.; Dalmas, E.; Thienel, C.; Dror, E.; Wiedemann, S.J.; et al. Inhibition of IL-1beta Improves Glycaemia in a Mouse Model for Gestational Diabetes. Sci. Rep. 2020, 10.
- Peng, H.Y.; Li, M.Q.; Li, H.P. High Glucose Suppresses the Viability and Proliferation of HTR-8/SVneo Cells through Regulation of the MiR-137/PRKAA1/IL-6 Axis. Int. J. Mol. Med. 2018, 42.
- Cawyer, C.; Afroze, S.H.; Drever, N.; Allen, S.; Jones, R.; Zawieja, D.C.; Kuehl, T.; Uddin, M.N. Attenuation of Hyperglycemia-Induced Apoptotic Signaling and Anti-Angiogenic Milieu in Cultured Cytotrophoblast Cells. Hypertens. Pregnancy 2016, 35.
- Rice, G.E.; Scholz-Romero, K.; Sweeney, E.; Peiris, H.; Kobayashi, M.; Duncombe, G.; Mitchell, M.D.; Salomon, C. The Effect of Glucose on the Release and Bioactivity of Exosomes from First Trimester Trophoblast Cells. J. Clin. Endocrinol. Metab. 2015, 100.
- Troncoso, F.; Acurio, J.; Herlitz, K.; Aguayo, C.; Bertoglia, P.; Guzman-Gutierrez, E.; Loyola, M.; Gonzalez, M.; Rezgaoui, M.; Desoye, G.; et al. Gestational Diabetes Mellitus Is Associated with Increased Pro-Migratory Activation of Vascular Endothelial Growth Factor Receptor 2 and Reduced Expression of Vascular Endothelial Growth Factor Receptor 1. PLoS ONE 2017, 12, e0182509.
- Loegl, J.; Nussbaumer, E.; Cvitic, S.; Huppertz, B.; Desoye, G.; Hiden, U. GDM Alters Paracrine Regulation of Feto-Placental Angiogenesis via the Trophoblast. Lab. Investig. 2017, 97.
- Landecho, M.F.; Tuero, C.; Valentí, V.; Bilbao, I.; de la Higuera, M.; Frühbeck, G. Relevance of Leptin and Other Adipokines in Obesity-Associated Cardiovascular Risk. Nutrients 2019, 11, 2664.
- Pérez-Pérez, A.; Maymó, J.; Gambino, Y.; Guadix, P.; Dueñas, J.L.; Varone, C.; Sánchez-Margalet, V. Insulin Enhances Leptin Expression in Human Trophoblastic Cells. Biol. Reprod. 2013, 89.
- Pérez-Pérez, A.; Toro, A.; Vilariño-García, T.; Maymó, J.; Guadix, P.; Dueñas, J.L.; Fernández-Sánchez, M.; Varone, C.; Sánchez-Margalet, V. Leptin Action in Normal and Pathological Pregnancies. J. Cell. Mol. Med. 2018, 22.
- Maymó, J.L.; Pérez Pérez, A.; Maskin, B.; Dueñas, J.L.; Calvo, J.C.; Sánchez Margalet, V.; Varone, C.L. The Alternative Epac/CAMP Pathway and the MAPK Pathway Mediate HCG Induction of Leptin in Placental Cells. PLoS ONE 2012, 7, e46216.
- Gambino, Y.P.; Pérez Pérez, A.; Dueñas, J.L.; Calvo, J.C.; Sánchez-Margalet, V.; Varone, C.L. Regulation of Leptin Expression by 17beta-Estradiol in Human Placental Cells Involves Membrane Associated Estrogen Receptor Alpha. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823.
- Magariños, M.P.; Sánchez-Margalet, V.; Kotler, M.; Calvo, J.C.; Varone, C.L. Leptin Promotes Cell Proliferation and Survival of Trophoblastic Cells. Biol. Reprod. 2007, 76.
- White, V.; González, E.; Capobianco, E.; Pustovrh, C.; Martínez, N.; Higa, R.; Baier, M.; Jawerbaum, A. Leptin Modulates Nitric Oxide Production and Lipid Metabolism in Human Placenta. Reprod. Fertil. Dev. 2006, 18.
- Côté, S.; Gagné-Ouellet, V.; Guay, S.P.; Allard, C.; Houde, A.A.; Perron, P.; Baillargeon, J.P.; Gaudet, D.; Guérin, R.; Brisson, D.; et al. PPARGC1α Gene DNA Methylation Variations in Human Placenta Mediate the Link between Maternal Hyperglycemia and Leptin Levels in Newborns. Clin. Epigenet. 2016, 8.
- Soheilykhah, S.; Mojibian, M.; Rahimi-Saghand, S.; Rashidi, M.; Hadinedoushan, H. Maternal Serum Leptin Concentration in Gestational Diabetes. Taiwan. J. Obstet. Gynecol. 2011, 50.
- Chen, D.; Xia, G.; Xu, P.; Dong, M. Peripartum Serum Leptin and Soluble Leptin Receptor Levels in Women with Gestational Diabetes. Acta Obstet. Gynecol. Scand. 2010, 89.
- Mokhtari, M.; Hashemi, M.; Yaghmaei, M.; Naderi, M.; Shikhzadeh, A.; Ghavami, S. Evaluation of the Serum Leptin in Normal Pregnancy and Gestational Diabetes Mellitus in Zahedan, Southeast Iran. Arch. Gynecol. Obstet. 2011, 284.
- Lepercq, J.; Cauzac, M.; Lahlou, N.; Timsit, J.; Girard, J.; Auwerx, J.; de Mouzon, S.H. Overexpression of Placental Leptin in Diabetic Pregnancy: A Critical Role for Insulin. Diabetes 1998, 47.
- Lappas, M.; Permezel, M.; Rice, G.E. Leptin and Adiponectin Stimulate the Release of Proinflammatory Cytokines and Prostaglandins from Human Placenta and Maternal Adipose Tissue via Nuclear Factor-ΚB, Peroxisomal Proliferator-Activated Receptor-γ and Extracellularly Regulated Kinase 1/2. Endocrinology 2005, 146.
- Cameo, P.; Bischof, P.; Calvo, J.C. Effect of Leptin on Progesterone, Human Chorionic Gonadotropin, and Interleukin-6 Secretion by Human Term Trophoblast Cells in Culture. Biol. Reprod. 2003, 68.
- Soh, E.B.E.; Mitchell, M.D.; Keelan, J.A. Does Leptin Exhibit Cytokine-like Properties in Tissues of Pregnancy? Am. J. Reprod. Immunol. 2000, 43.
- Miehle, K.; Stepan, H.; Fasshauer, M. Leptin, Adiponectin and Other Adipokines in Gestational Diabetes Mellitus and Pre-Eclampsia. Clin. Endocrinol. 2012, 76.
- De Gennaro, G.; Palla, G.; Battini, L.; Simoncini, T.; del Prato, S.; Bertolotto, A.; Bianchi, C. The Role of Adipokines in the Pathogenesis of Gestational Diabetes Mellitus. Gynecol. Endocrinol. 2019, 35.
- Retnakaran, R.; Qi, Y.; Connelly, P.W.; Sermer, M.; Hanley, A.J.; Zinman, B. Low Adiponectin Concentration during Pregnancy Predicts Postpartum Insulin Resistance, Beta Cell Dysfunction and Fasting Glycaemia. Diabetologia 2010, 53.
- Williams, M.A.; Qiu, C.; Muy-Rivera, M.; Vadachkoria, S.; Song, T.; Luthy, D.A. Plasma Adiponectin Concentrations in Early Pregnancy and Subsequent Risk of Gestational Diabetes Mellitus. J. Clin. Endocrinol. Metab. 2004, 89.
- Ramachandrayya, S.A.; D’Cunha, P.; Rebeiro, C. Maternal Circulating Levels of Adipocytokines and Insulin Resistance as Predictors of Gestational Diabetes Mellitus: Preliminary Findings of a Longitudinal Descriptive Study. J. Diabetes Metab. Disord. 2020, 19.
- Chen, J.; Tan, B.; Karteris, E.; Zervou, S.; Digby, J.; Hillhouse, E.W.; Vatish, M.; Randeva, H.S. Secretion of Adiponectin by Human Placenta: Differential Modulation of Adiponectin and Its Receptors by Cytokines. Diabetologia 2006, 49.
- Benaitreau, D.; dos Santos, E.; Leneveu, M.C.; Alfaidy, N.; Feige, J.J.; de Mazancourt, P.; Pecquery, R.; Dieudonné, M.N. Effects of Adiponectin on Human Trophoblast Invasion. J. Endocrinol. 2010, 207.
- Bouchard, L.; Hivert, M.F.; Guay, S.P.; St-Pierre, J.; Perron, P.; Brisson, D. Placental Adiponectin Gene DNA Methylation Levels Are Associated with Mothers’ Blood Glucose Concentration. Diabetes 2012, 61.
- Dias, S.; Adam, S.; Abrahams, Y.; Rheeder, P.; Pheiffer, C. Adiponectin DNA Methylation in South African Women with Gestational Diabetes Mellitus: Effects of HIV Infection. PLoS ONE 2021, 16, e0248694.
- King, A.E.; Paltoo, A.; Kelly, R.W.; Sallenave, J.M.; Bocking, A.D.; Challis, J.R.G. Expression of Natural Antimicrobials by Human Placenta and Fetal Membranes. Placenta 2007, 28.
- Yarbrough, V.L.; Winkle, S.; Herbst-Kralovetz, M.M. Antimicrobial Peptides in the Female Reproductive Tract: A Critical Component of the Mucosal Immune Barrier with Physiological and Clinical Implications. Hum. Reprod. Update 2015, 21.
- Zaga-Clavellina, V.; Garcia-Lopez, G.; Flores-Espinosa, P. Evidence of in Vitro Differential Secretion of Human Beta-Defensins-1, -2, and -3 after Selective Exposure to Streptococcus Agalactiae in Human Fetal Membranes. J. Matern. Fetal Neonatal Med. 2012, 25.
- Zaga-Clavellina, V.; Martha, R.V.M.; Flores-Espinosa, P. In Vitro Secretion Profile of Pro-Inflammatory Cytokines IL-1β, TNF-α, IL-6, and of Human Beta-Defensins (HBD)-1, HBD-2, and HBD-3 from Human Chorioamniotic Membranes after Selective Stimulation with Gardnerella Vaginalis. Am. J. Reprod. Immunol. 2012, 67.
- Liu, N.; Kaplan, A.T.; Low, J.; Nguyen, L.; Liu, G.Y.; Equils, O.; Hewison, M. Vitamin D Induces Innate Antibacterial Responses in Human Trophoblasts via an Intracrine Pathway 1. Biol. Reprod. 2009, 80.
- Klaffenbach, D.; Friedrich, D.; Strick, R.; Strissel, P.L.; Beckmann, M.W.; Rascher, W.; Gessner, A.; Dötsch, J.; Meißner, U.; Schnare, M. Contribution of Different Placental Cells to the Expression and Stimulation of Antimicrobial Proteins (AMPs). Placenta 2011, 32.
- Svinarich, D.M.; Gomez, R.; Romero, R. Detection of Human Defensins in the Placenta. Am. J. Reprod. Immunol. 1997, 38.
- Kim, H.S.; Cho, J.H.; Park, H.W.; Yoon, H.; Kim, M.S.; Kim, S.C. Endotoxin-Neutralizing Antimicrobial Proteins of the Human Placenta. J. Immunol. 2002, 168.
- Oliveira-Bravo, M.; Sangiorgi, B.B.; Schiavinato, J.L.D.S.; Carvalho, J.L.; Covas, D.T.; Panepucci, R.A.; Neves, F.D.A.R.; Franco, O.L.; Pereira, R.W.; Saldanha-Araujo, F. LL-37 Boosts Immunosuppressive Function of Placenta-Derived Mesenchymal Stromal Cells. Stem Cell Res. Ther. 2016, 7.
- Szukiewicz, D.; Alkhalayla, H.; Pyzlak, M.; Szewczyk, G. High Glucose Culture Medium Downregulates Production of Human β-Defensin-2 (HBD-2) in Human Amniotic Epithelial Cells (HAEC). FASEB J. 2016, 30, 307. Available online: https://faseb.onlinelibrary.wiley.com/doi/abs/10.1096/fasebj.30.1_supplement.307.1?cited-by=yes&legid=fasebj%3B30%2F1_Supplement%2F307.1 (accessed on 1 June 2021).
- Montoya-Rosales, A.; Castro-Garcia, P.; Torres-Juarez, F.; Enciso-Moreno, J.A.; Rivas-Santiago, B. Glucose Levels Affect LL-37 Expression in Monocyte-Derived Macrophages Altering the Mycobacterium Tuberculosis Intracellular Growth Control. Microb. Pathog. 2016, 97.
- Froy, O.; Hananel, A.; Chapnik, N.; Madar, Z. Differential Effect of Insulin Treatment on Decreased Levels of Beta-Defensins and Toll-like Receptors in Diabetic Rats. Mol. Immunol. 2007, 44.
- Rivas-Santiago, B.; Trujillo, V.; Montoya, A.; Gonzalez-Curiel, I.; Castañeda-Delgado, J.; Cardenas, A.; Rincon, K.; Hernandez, M.L.; Hernández-Pando, R. Expression of Antimicrobial Peptides in Diabetic Foot Ulcer. J. Dermatol. Sci. 2012, 65.
- Olmos-Ortiz, A.; García-Quiroz, J.; Avila, E.; Caldiño-Soto, F.; Halhali, A.; Larrea, F.; Díaz, L. Lipopolysaccharide and CAMP Modify Placental Calcitriol Biosynthesis Reducing Antimicrobial Peptides Gene Expression. Am. J. Reprod. Immunol. 2018, 79.
More