Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Peter Tang and Version 1 by Barry Wilk.
Modified citrus pectin (MCP) has a low-molecular-weight degree of esterification to allow absorption from the small intestinal epithelium into the circulation. MCP produces pleiotropic effects, including but not limited to its antagonism of galectin-3, which have shown benefit in preclinical and clinical models. Regarding cancer, MCP modulates several rate-limiting steps of the metastatic cascade. MCP can also affect cancer cell resistance to chemotherapy. Regarding fibrotic diseases, MCP modulates many of the steps involved in the pathogenesis of aortic stenosis. MCP also reduces fibrosis to the kidney, liver, and adipose tissue. Other benefits of MCP include detoxification and improved immune function.
- cancer
- cardiovascular
- fibrosis
- galectin
- inflammation
- pectasol
1. Introduction
Citrus pectin is a soluble dietary fiber derived from the white pith of citrus fruit peels. Pectin is a large and complex molecule in its natural form, weighing 60–300 kilodalton (kDa), and containing a variable degree of (as much as ~70%) esterification. Pectins are a family of covalently linked galacturonic acid-rich polymers, with three identified central pectic polysaccharides regions: homogalacturonan (HG), rhamnogalacturonan-I (RG-I), and substituted galacturonans (GS). Among the GS is rhamnogalacturonan-II (RG-II), which is distinct from RG-I. RG-II has four types of structurally different oligosaccharides chains composed of 12 kinds of glycosyl residues [1] (Figure 1). Native pectin is not degraded during human digestion, and its large size prevents intestinal absorption [2,3][2][3]. However, when citrus pectin is modified (MCP) with a specific pH and heat-controlled enzymatic treatment to yield a product with a low molecular weight of <15 kilodaltons (kDa) and a degree of esterification under 5%, it can be absorbed from the small intestinal epithelium into the circulation [3].
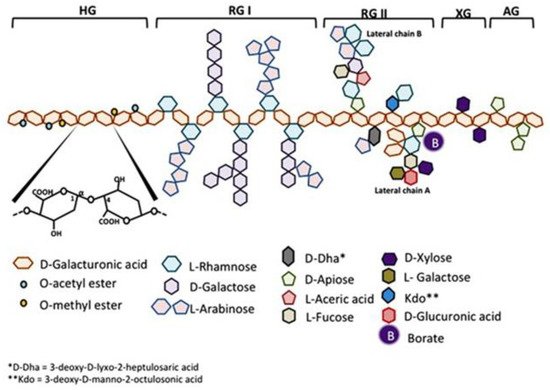
Figure 1. Schematic representation of pectin structure. AG, arabinogalactan; HG, homogalacturonan; RG, rhamnogalacturonan; XG, xylogalacturonan. It is reproduced with permission under a Creative Commons Attribution License (CC BY 4.0) https://creativecommons.org/licenses/by/4.0/ from Leclere, L.; Cutsem, PV.; Michiels, C.; Anti-cancer activities of pH- or heat-modified pectin. Front Pharmacol. 2013 Oct 8; 4:128 [1].
2. Galectin-3
Most biomedical reports of MCP focus specifically on its antagonism of galectin-3 (Gal-3). Located in the nucleus, cytoplasm, outer cell surface, and extracellular space, Gal-3 is a member of a β-galactoside-binding lectin family [49][4]. Galectin-3 is a unique chimeric galectin containing a single carbohydrate recognition domain (CRD) of 130 amino acids at the COOH terminal with a unique 12 amino acid NH2-terminal domain (NTD). The CRD also contains what has been referred to as the anti-death motif or Asp-Trp-Gly-Arg (NWGR) due to sequence similarity to anti-apoptotic B-cell lymphoma-2 (Bcl-2) protein [50][5]. In the NTD, the serine 6 can be phosphorylated by casein kinases 1 and 2, contributing to nuclear translocation and a reduction of affinity to its ligands. Connecting the CRD and the NTD is a collagen-like sequence (CLS) domain consisting of about 100 amino acids and contains a Pro-Gly-Ala-Tyr-rich repeat motif. This long tail allows for unique pentameric oligomerization and contains the collagenase cleavable H-domain (Figure 2) [51][6]. Galectin-3 plays a prominent role in the progression of cancer and fibrotic diseases [52][7].
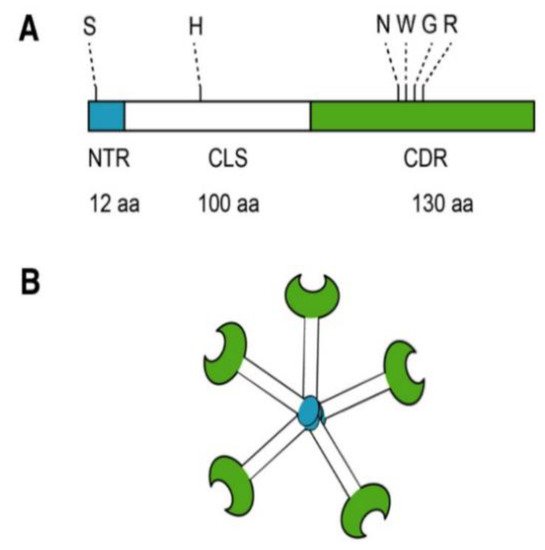
Figure 2. Structure of Gal-3. (A) Gal-3 protein structure consists of an N terminal Domain (NTD), which has an N terminal region of 12 amino acids (aa) and contains a serine 6 (S) phosphorylation site. The carbohydrate recognition domain (CRD) of 130 aa comprises the C-terminal and contains the anti-death motif or Asp-Trp-Gly-Arg (NWGR); (B) Pentameric structure of Gal-3. Reproduced with permission under a Creative Commons Attribution License (CC BY 4.0) https://creativecommons.org/licenses/by/4.0/. Clementy, N.; Piver, E.; Bisson, A.; André, Clémentine, A.; Bernard, A.; Pierre, B.; Fauchier, L.; Babuty, D. (2018). Galectin-3 in Atrial Fibrillation: Mechanisms and Therapeutic Implications. Int J Mol Sci. 2018 Apr; 19(4): 976 [49][4].
Regarding the former, the damaging effects of Gal-3 are due to its ability to accelerate the rate-limiting steps of metastasis [53][8]. Regarding the latter, the detrimental effects are due to its ability to bind matrix proteins such as cell surface receptors (integrins), collagen, elastin, and fibronectin and form cross-linking lattices in the extracellular matrix (ECM) [54][9]. Modified citrus pectin is abundant in β-galactose [53][8], which allows it to bind tightly to Gal-3 and modulate its bioactivity [55][10].
3. Cancer
Most of the morbidity and mortality associated with cancer is caused by metastasis, which is the migration of cancer from the site of primary tumor growth to distant organs and tissues. The metastatic cascade contains several rate-limiting steps that are modulated by Gal-3 and, in turn, by MCP as well [53][8]. The first step for neoplastic cells is to survive apoptosis that is associated with the loss of anchorage (anoikis) following escape from the primary tumor and intravasation. Galectin-3 protects cancer cells from anoikis [56,57][11][12] by causing a cell cycle arrest at the late G1 phase, which is an anoikis-insensitive point [56][11]. MCP has been shown to downregulate cyclin B and cdc2 in human prostatic JCA-1 cells [58][13], which may cause an accumulation of cancer cells in G2/M, thereby inducing apoptosis.
The next rate-limiting step in metastasis involves tumor cell arrest in distant organ microvasculature. Galectin-3 has been shown to mediate metastatic cell adhesion to the endothelium [59,60,61,62,63][14][15][16][17][18]. MCP was demonstrated to inhibit tumor cell adhesion to the endothelium as well as cancer cell homotypic aggregation involved in metastatic cell arrest in distant organs and the formation of intravascular metastatic deposits [59,64,65,66,67,68][14][19][20][21][22][23].
The third rate-limiting step in metastasis involves a forking point where tumor cells can either proliferate inside organ microvessels until the metastatic tumor outgrows the blood vessel and invades distant organ parenchyma [69][24], or extravasate before starting secondary tumor growth. Invasive propensity involves a series of tumor cell interactions with ECM proteins associated with the basement membrane and target organ stroma. MCP has been shown to reduce Gal-3-mediated tumor cell interactions with ECM proteins such as laminin [66][21]. Also, citrus pectin polysaccharides dose-dependently decreased the invasion through matrigel of human endothelial cells [67][22], of MDA-MB-231 human metastatic breast carcinoma cells [70][25], and human buccal metastatic cells [70][25].
4. Fibrotic Diseases
4.1. Aortic Stenosis
Many of the steps involved in the pathogenesis of aortic stenosis (AS) are modulated by Gal-3 and, therefore, presumably also by MCP [88][26]. Galectin-3 transforms quiescent fibroblasts into myofibroblasts that produce and secrete matrix proteins such as collagen [89,90][27][28]. Galectin-3 also affects collagen maturation and cross-linking [91,92][29][30]. It also stimulates pro-inflammatory mediators [93][31]. In human cardiac fibroblasts, Gal-3 increases the production and secretion of interleukin (IL)-1β, IL-6, monocyte chemoattractant protein-1, collagen type I and type III, and fibronectin [31][32]. It also increases the activity of metalloproteinases-1, -2, and -9 [31][32]. Treatment with MCP abrogates these effects. In the vasculature, Gal-3 promotes arterial stiffness by enhancing the production and secretion of pro-fibrotic and pro-inflammatory markers in vascular smooth muscle cells [33]. Again, MCP reverses this effect. In endothelial cells, Gal-3 enhances the expression of inflammatory factors, chemokines, and adhesion molecules [94][34]. Galectin-3 also affects the cell surface expression and activation of vascular endothelial growth factor receptor 2 in human endothelial cells, which contributes to plasma membrane retention and promotes angiogenesis [95][35]. In interstitial valve cells from the aorta, Gal-3 enhances the secretion of inflammatory and fibrotic mediators and increases the expression of calcification mediators [29][36]. The administration of MCP prevents these effects. MCP also prevented an increase in cardiac Gal-3, and normalized histological and molecular alterations in short-term AS [27][37].
4.2. Additional Cardiovascular Effects
Galectin-3 inhibition with MCP prevented cardiac inflammation and fibrosis associated with an excess of aldosterone levels independently of blood pressure levels [96][38]. Galectin-3 antagonism with MCP and aldosterone opposition reversed isoproterenol-induced left ventricular systolic dysfunction, thereby preventing the development of myocardial fibrosis in this mice model with selective cardiac hyperaldosteronism [30][39]. Blockade by MCP in a pressure overload-induced model, which increases Gal-3, showed amelioration of media thickness, fibrosis, and inflammation in aortic valve calcification [28][40]. It also reduced the size of atherosclerotic lesion areas by inhibiting the adhesion of leukocytes to endothelial cells [26][41].
Furthermore, MCP inhibited Gal-3 and decreased experimental abdominal aortic aneurysm development [25][42]. MCP reduced cardiac lipotoxicity and ameliorated cardiac mitochondrial damage in an obesity model [24][43]. Also, MCP restored the levels of cardiac peroxiredoxin-4 as well as prohibitin-2 levels and improved oxidative status [23][44]. Neurological impairments were prevented with MCP on post-aneurysmal subarachnoid hemorrhage, suggesting pleiotropic neuroprotective action, such as anti-neuroinflammatory and anti-apoptotic effects, beyond protecting the blood-brain barrier through inhibiting Gal-3 [22][45]. Inhibition mediated by the MCP of Gal-3 in mice prevented the pro-fibrotic and pro-inflammatory effects of cardiotrophin-1 [21][46]. Furthermore, MCP and perindopril comparably improved ischemic heart failure by downregulating Gal-3 and reducing myocardial fibrosis [20][47]. The blockade of Gal-3 with MCP prevented cardiac fibrosis, inflammation, and functional alterations [19][48].
4.3. Kidney
In a model of experimental acute kidney injury, all folic acid-treated mice lost weight while their kidneys enlarged secondary to the renal insult; MCP significantly lessened these gross changes, but this was not associated with changes in Gal-3 expression [36][49]. Via its blocking action on Gal-3, MCP protected against aldosterone-induced cardiac and renal fibrosis and dysfunction [32][50]. The inhibition of Gal-3 normalized renal Gal-3 levels as well as functional, histological, and molecular alterations in an obese and AS model, preventing renal fibrosis, inflammation, and damage with MCP treatment [35][51]. Moreover, MCP attenuated early renal impairment in spontaneously hypertensive rats as indicated by reduced albuminuria, improved renal function, and decreased renal fibrosis, epithelial-mesenchymal transition, and inflammation [34][52].
4.4. Additional Fibrotic Diseases
In a model of diet-induced obesity, MCP prevented adipose tissue fibrosis, inflammation, and the increase in adipocyte differentiation markers despite not affecting body weight, adipose tissue weights, or adiposity [37][53]. Liver fibrosis was alleviated with MCP and aided in hepatic regeneration, which may be mediated by an antioxidant effect [38][54].
5. Detoxification
To date, MCP has been used in four clinical studies of detoxification. Treatment with MCP increased urinary excretion of lead, arsenic, and cadmium in healthy volunteers, without side effects or depletion of essential elements [42][55]. In a case study of five patients, there was an average of 74% reduction in lead or mercury without side effects with the use of MCP alone or with a MCP/alginates combination [41][56]. Treatment with MCP dramatically decreased the levels of lead in blood and increased the levels of lead in urine in children hospitalized with lead toxicity [40][57]. Fecal uranium excretion was promoted by MCP/alginate supplement without side effects in a family with low-level chronic exposure from their environment and diet [39][58].
6. Immune Function
There are many reports of various effects on immunity with MCP. There was significant activation of T-cytotoxic and natural killer (NK) cells in blood cultures by MCP, and the NK-cells demonstrated functionality against K562 leukemic cells in culture. The presence of a low degree of methyl esterification and flexible low-molecular-weight pectin polymer enriched in saturated and unsaturated oligogalacturonic acids appear to be the immunostimulatory carbohydrates in MCP [48][59]. An additive effect of MCP in combination with cefotaxime against all six methicillin-resistant Staphylococcus aureus (MRSA) strains has been shown [47][60]. Honokiol, a purified extract from magnolia bark used in traditional Asian medicine and MCP, has been shown to have synergistic antioxidant activity and anti-inflammatory effects [46][61]. There was an inhibition of toxin-producing Escherichia coli adhesion and reduced Shiga toxin cytotoxicity with MCP [45][62]. Furthermore, MCP co-administration with live probiotic L. acidophilus ATCC 4356 supplement helped maintain or improve the integrity and population of the intestinal microbiota [44][63]. Finally, MCP has an immunomodulatory effect on the levels of cytokine secretion in the spleen of mice, which may be regulated by IL-4 [43][64].
7. Other Galectin-3 Inhibitors
There are other laboratory MCPs prepared by just heat and pH treatment. Heat by autoclaving MCP induced cell death in HepG2 and A549 cells. The induced cell death was different from classical apoptosis because there was no DNA cleavage [97][65]. Also, the delivery of autoclaved MCP reduced plaque volume in apolipoprotein E-deficient mice [98][66]. Renal cell carcinoma cells cotreated with autoclaved pectin and arsenic trioxide demonstrated increased apoptosis [99][67]. Synergistic treatment with S-trans, transfarnesylthiosalicylic acid and pH modified citrus pectin inhibited anaplastic thyroid cells proliferation in vitro by inducing cell cycle arrest and increased apoptosis rate [100][68]. pH modified citrus pectin also reduced the growth of solid tumors in balb-c mice [57][12].
Several Gal-3 inhibitors are also in pharmaceutical development. An injectable MCP in pharmaceutical development now abandoned called GCS-100 induced apoptosis in acute myeloid leukemia cells [101][69]. It also removed cell-surface Gal-3 from CD45, thus rendering diffuse large B-cell lymphoma cells susceptible to chemotherapeutic agents [102][70]. GCS-100 also detached Gal-3 from tumor-infiltrating lymphocytes and improved the cytotoxicity and secretion of different cytokines [103][71]. Furthermore, GCS-100 induced the inhibition of proliferation, the accumulation of cells in sub-G1 and G1 phases, and apoptosis with the activation of both the caspase-8 and -9 pathways [104][72]. GCS-100 enhanced calpain activation, which reduced the proapoptotic effect of Gal-3 [105][73].
Another Gal-3 inhibitor in pharmaceutical development is an inhalable formulation called TD139, a thiodigalactoside derivative. This inhibitor abrogated the susceptibility to natural killer T-cell-dependent hepatitis [106][74]. Pretreatment of wild-type C57BL/6 mice with TD139 lowered liver injury and led to milder infiltration of interferon-gamma and interleukin (IL)-17 and -4-producing cluster of differentiation (CD)4(+) T cells, and an increase in the total number of IL-10-producing CD4(+) T cells and F4/80(+) CD206(+) activating macrophages, and prevented the apoptosis of liver-infiltrating mononuclear cells [107][75]. TD139 blocked transforming growth factor-β-induced β-catenin activation in vitro and in vivo and lowered the late-stage progression of lung fibrosis after treatment with bleomycin [108][76].
Other pectin carbohydrate-based galectin inhibitors in drug development, injectables GR-MD-02 (galactoarabino-rhamnogalacturonan) and GM-CT-01 (galactomannan) resulted in a lowering of fibrosis with the reduction in the portal and septal Gal-3 positive macrophages and reductions in portal pressure [109][77]. The treatment resulted in sharp improvement in liver histology, with a significant decrease in non-alcoholic steatohepatitis (NASH) activity and collagen deposition; GM-CT-01 had an intermediate effect between the vehicle and GR-MD-02 [110][78].
References
- Leclere, L.; Van Cutsem, P.; Michiels, C. Anti-cancer activities of pH-or heat-modified pectin. Front. Pharm. 2013, 4, 128.
- Niture, S.K.; Refai, L. Plant pectin: A potential source for cancer suppression. Am. J. Pharm. Toxicol. 2013, 8, 9–19.
- Courts, F.L. Profiling of modified citrus pectin oligosaccharide transport across Caco-2 cell monolayers. PharmaNutrition 2013, 1, 22–31.
- Dumic, J.; Dabelic, S.; Flögel, M. Galectin-3: An open-ended story. Biochim. Biophys. Acta 2006, 1760, 616–635.
- Harazono, Y.; Nakajima, K.; Raz, A. Why anti-Bcl-2 clinical trials fail: A solution. Cancer Metastasis Rev. 2014, 33, 285–294.
- Clementy, N.; Piver, E.; Bisson, A.; Andre, C.; Bernard, A.; Pierre, B.; Fauchier, L.; Babuty, D. Galectin-3 in atrial fibrillation: Mechanisms and therapeutic implications. Int. J. Mol. Sci. 2018, 19, 976.
- Dong, R.; Zhang, M.; Hu, Q.; Zheng, S.; Soh, A.; Zheng, Y.; Yuan, H. Galectin-3 as a novel biomarker for disease diagnosis and a target for therapy. Int. J. Mol. Med. 2018, 41, 599–614.
- Glinsky, V.V.; Raz, A. Modified citrus pectin anti-metastatic properties: One bullet, multiple targets. Carbohydr. Res. 2009, 344, 1788–1791.
- Fortuna-Costa, A.; Gomes, A.M.; Kozlowski, E.O.; Stelling, M.P.; Pavão, M.S. Extracellular galectin-3 in tumor progression and metastasis. Front. Oncol. 2014, 4, 138.
- Gao, X.; Zhi, Y.; Zhang, T.; Xue, H.; Wang, X.; Foday, A.D.; Tai, G.; Zhou, Y. Analysis of the neutral polysaccharide fraction of MCP and its inhibitory activity on galectin-3. Glycoconj. J. 2012, 29, 159–165.
- Kim, H.-R.C.; Lin, H.-M.; Biliran, H.; Raz, A. Cell cycle arrest and inhibition of anoikis by galectin-3 in human breast epithelial cells. Cancer Res. 1999, 59, 4148–4154.
- Hayashi, A.; Gillen, A.C.; Lott, J.R. Effects of daily oral administration of quercetin chalcone and modified citrus pectin on implanted colon-25 tumor growth in Balb-c mice. Altern. Med. Rev. 2000, 5, 546–552.
- Hsieh, T.; Wu, J.M. Changes in cell growth, cyclin/kinase, endogenous phosphoproteins and nm23 gene expression in human prostatic JCA-1 cells treated with modified citrus pectin. Biochem. Mol. Biol. Int. 1995, 37, 833–841.
- Lehr, J.E.; Pienta, K.J. Preferential adhesion of prostate cancer cells to a human bone marrow endothelial cell line. J. Natl. Cancer Inst. 1998, 90, 118–123.
- Glinsky, V.V.; Glinsky, G.V.; Rittenhouse-Olson, K.; Huflejt, M.E.; Glinskii, O.V.; Deutscher, S.L.; Quinn, T.P. The role of Thomsen-Friedenreich antigen in adhesion of human breast and prostate cancer cells to the endothelium. Cancer Res. 2001, 61, 4851–4857.
- Khaldoyanidi, S.K.; Glinsky, V.V.; Sikora, L.; Glinskii, A.B.; Mossine, V.V.; Quinn, T.P.; Glinsky, G.V.; Sriramarao, P. MDA-MB-435 human breast carcinoma cell homo-and heterotypic adhesion under flow conditions is mediated in part by Thomsen-Friedenreich antigen-galectin-3 interactions. J. Biol. Chem. 2003, 278, 4127–4134.
- Glinsky, V.V.; Glinsky, G.V.; Glinskii, O.V.; Huxley, V.H.; Turk, J.R.; Mossine, V.V.; Deutscher, S.L.; Pienta, K.J.; Quinn, T.P. Intravascular metastatic cancer cell homotypic aggregation at the sites of primary attachment to the endothelium. Cancer Res. 2003, 63, 3805–3811.
- Glinskii, O.V.; Turk, J.R.; Pienta, K.J.; Huxley, V.H.; Glinsky, V.V. Evidence of porcine and human endothelium activation by cancer-associated carbohydrates expressed on glycoproteins and tumour cells. J. Physiol. 2004, 554, 89–99.
- Platt, D.; Raz, A. Modulation of the lung colonization of B16-F1 melanoma cells by citrus pectin. J. Natl. Cancer Inst. 1992, 84, 438–442.
- Pienta, K.J.; Nailk, H.; Akhtar, A.; Yamazaki, K.; Replogle, T.S.; Lehr, J.; Donat, T.L.; Tait, L.; Hogan, V.; Raz, A. Inhibition of spontaneous metastasis in a rat prostate cancer model by oral administration of modified citrus pectin. J. Natl. Cancer Inst. 1995, 87, 348–353.
- Inohara, H.; Raz, A. Effects of natural complex carbohydrate (citrus pectin) on murine melanoma cell properties related to galectin-3 functions. Glycoconj. J. 1994, 11, 527–532.
- Nangia-Makker, P.; Hogan, V.; Honjo, Y.; Baccarini, S.; Tait, L.; Bresalier, R.; Raz, A. Inhibition of human cancer cell growth and metastasis in nude mice by oral intake of modified citrus pectin. J. Natl. Cancer Inst. 2002, 94, 1854–1862.
- Glinskii, O.V.; Huxley, V.H.; Glinsky, G.V.; Pienta, K.J.; Raz, A.; Glinsky, V.V. Mechanical entrapment is insufficient and intercellular adhesion is essential for metastatic cell arrest in distant organs. Neoplasia 2005, 7, 522–527.
- Al-Mehdi, A.; Tozawa, K.; Fisher, A.; Shientag, L.; Lee, A.; Muschel, R. Intravascular origin of metastasis from the proliferation of endothelium-attached tumor cells: A new model for metastasis. Nat. Med. 2000, 6, 100–102.
- Sathisha, U.; Jayaram, S.; Nayaka, M.H.; Dharmesh, S.M. Inhibition of galectin-3 mediated cellular interactions by pectic polysaccharides from dietary sources. Glycoconj. J. 2007, 24, 497–507.
- Arrieta, V.; Sádaba, J.; Álvarez, V.; Rodríguez, J.; López-Andrés, N. Galectin-3 as a novel biotarget in cardiovascular alterations associated to development of severe aortic stenosis. Sist. Sanit. Navar. 2019, 72347.
- Sharma, U.C.; Pokharel, S.; van Brakel, T.J.; van Berlo, J.H.; Cleutjens, J.P.; Schroen, B.; André, S.; Crijns, H.J.; Gabius, H.-J.; Maessen, J. Galectin-3 marks activated macrophages in failure-prone hypertrophied hearts and contributes to cardiac dysfunction. Circulation 2004, 110, 3121–3128.
- Yu, L.; Ruifrok, W.P.; Meissner, M.; Bos, E.M.; van Goor, H.; Sanjabi, B.; van der Harst, P.; Pitt, B.; Goldstein, I.J.; Koerts, J.A. Genetic and pharmacological inhibition of galectin-3 prevents cardiac remodeling by interfering with myocardial fibrogenesis. Circ. Heart Fail. 2013, 6, 107–117.
- Ho, M.-K.; Springer, T. Mac-2, a novel 32,000 Mr mouse macrophage subpopulation-specific antigen defined by monoclonal antibodies. J. Immunol. 1982, 128, 1221–1228.
- De Boer, R.A.; Van Der Velde, A.R.; Mueller, C.; Van Veldhuisen, D.J.; Anker, S.D.; Peacock, W.F.; Adams, K.F.; Maisel, A. Galectin-3: A modifiable risk factor in heart failure. Cardiovasc. Drugs 2014, 28, 237–246.
- Papaspyridonos, M.; McNeill, E.; de Bono, J.P.; Smith, A.; Burnand, K.G.; Channon, K.M.; Greaves, D.R. Galectin-3 is an amplifier of inflammation in atherosclerotic plaque progression through macrophage activation and monocyte chemoattraction. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 433–440.
- Martínez-Martínez, E.; López-Ándres, N.; Jurado-López, R.; Rousseau, E.; Bartolomé, M.V.; Fernández-Celis, A.; Rossignol, P.; Islas, F.; Antequera, A.; Prieto, S. Galectin-3 participates in cardiovascular remodeling associated with obesity. Hypertension 2015, 66, 961–969.
- Calvier, L.; Miana, M.; Reboul, P.; Cachofeiro, V.; Martinez-Martinez, E.; de Boer, R.A.; Poirier, F.; Lacolley, P.; Zannad, F.; Rossignol, P. Galectin-3 mediates aldosterone-induced vascular fibrosis. Arter. Thromb. Vasc. Biol. 2013, 33, 67–75.
- Chen, X.; Zhang, R.; Zhang, Q.; Xu, Z.; Xu, F.; Li, D.; Li, Y. Microtia patients: Auricular chondrocyte ECM is promoted by CGF through IGF-1 activation of the IGF-1R/PI3K/AKT pathway. J. Cell. Physiol. 2019, 234, 21817–21824.
- Markowska, A.I.; Jefferies, K.C.; Panjwani, N. Galectin-3 protein modulates cell surface expression and activation of vascular endothelial growth factor receptor 2 in human endothelial cells. J. Biol. Chem. 2011, 286, 29913–29921.
- Sádaba, J.R.; Martínez-Martínez, E.; Arrieta, V.; Álvarez, V.; Fernández-Celis, A.; Ibarrola, J.; Melero, A.; Rossignol, P.; Cachofeiro, V.; López-Andrés, N. Role for galectin-3 in calcific aortic valve stenosis. J. Am. Heart Assoc. 2016, 5, e004360.
- Arrieta, V.; Martinez-Martinez, E.; Ibarrola, J.; Alvarez, V.; Sádaba, R.; Garcia-Peña, A.; Fernández-Celis, A.; Cachofeiro, V.; Rossignol, P.; López-Andrés, N. A role for galectin-3 in the development of early molecular alterations in short-term aortic stenosis. Clin. Sci. 2017, 131, 935–949.
- Martínez-Martínez, E.; Calvier, L.; Fernández-Celis, A.; Rousseau, E.; Jurado-López, R.; Rossoni, L.V.; Jaisser, F.; Zannad, F.; Rossignol, P.; Cachofeiro, V. Galectin-3 blockade inhibits cardiac inflammation and fibrosis in experimental hyperaldosteronism and hypertension. Hypertension 2015, 66, 767–775.
- Vergaro, G.; Prud’homme, M.; Fazal, L.; Merval, R.; Passino, C.; Emdin, M.; Samuel, J.-L.; Cohen Solal, A.; Delcayre, C. Inhibition of galectin-3 pathway prevents isoproterenol-induced left ventricular dysfunction and fibrosis in mice. Hypertension 2016, 67, 606–612.
- Ibarrola, J.; Martínez-Martínez, E.; Sádaba, J.; Arrieta, V.; García-Peña, A.; Álvarez, V.; Fernández-Celis, A.; Gainza, A.; Rossignol, P.; Cachofeiro Ramos, V. Beneficial effects of galectin-3 blockade in vascular and aortic valve alterations in an experimental pressure overload model. Int. J. Mol. Sci. 2017, 18, 1664.
- Lu, Y.; Zhang, M.; Zhao, P.; Jia, M.; Liu, B.; Jia, Q.; Guo, J.; Dou, L.; Li, J. Modified citrus pectin inhibits galectin-3 function to reduce atherosclerotic lesions in apoE-deficient mice. Mol. Med. Rep. 2017, 16, 647–653.
- Fernandez-García, C.-E.; Tarin, C.; Roldan-Montero, R.; Martinez-Lopez, D.; Torres-Fonseca, M.; Lindhot, J.S.; de Ceniga, M.V.; Egido, J.; Lopez-Andres, N.; Blanco-Colio, L.-M. Increased galectin-3 levels are associated with abdominal aortic aneurysm progression and inhibition of galectin-3 decreases elastase-induced AAA development. Clin. Sci. 2017, 131, 2707–2719.
- Marín-Royo, G.; Gallardo, I.; Martínez-Martínez, E.; Gutiérrez, B.; Jurado-López, R.; López-Andrés, N.; Gutiérrez-Tenorio, J.; Rial, E.; Bartolomé, M.a.V.; Nieto, M.L. Inhibition of galectin-3 ameliorates the consequences of cardiac lipotoxicity in a rat model of diet-induced obesity. Dis. Model. Mech. 2018, 11, dmm032086.
- Ibarrola, J.; Arrieta, V.; Sádaba, R.; Martinez-Martinez, E.; Garcia-Peña, A.; Alvarez, V.; Fernández-Celis, A.; Gainza, A.; Santamaría, E.; Fernández-Irigoyen, J. Galectin-3 down-regulates antioxidant peroxiredoxin-4 in human cardiac fibroblasts: A new pathway to induce cardiac damage. Clin. Sci. 2018, 132, 1471–1485.
- Nishikawa, H.; Liu, L.; Nakano, F.; Kawakita, F.; Kanamaru, H.; Nakatsuka, Y.; Okada, T.; Suzuki, H. Modified citrus pectin prevents blood-brain barrier disruption in mouse subarachnoid hemorrhage by inhibiting galectin-3. Stroke 2018, 49, 2743–2751.
- Martínez-Martínez, E.; Brugnolaro, C.; Ibarrola, J.; Ravassa, S.; Buonafine, M.; López, B.; Fernández-Celis, A.; Querejeta, R.; Santamaria, E.; Fernández-Irigoyen, J. CT-1 (Cardiotrophin-1)-Gal-3 (Galectin-3) Axis in Cardiac Fibrosis and Inflammation: Mechanistic Insights and Clinical Implications. Hypertension 2019, 73, 602–611.
- Li, S.; Li, S.; Hao, X.; Zhang, Y.; Deng, W. Perindopril and a galectin-3 inhibitor improve ischemic heart failure in rabbits by reducing Gal-3 expression and myocardial fibrosis. Front. Physiol. 2019, 10, 267.
- Ibarrola, J.; Matilla, L.; Martínez-Martínez, E.; Gueret, A.; Fernández-Celis, A.; Henry, J.-P.; Nicol, L.; Jaisser, F.; Mulder, P.; Ouvrard-Pascaud, A. Myocardial Injury After Ischemia/Reperfusion Is Attenuated By Pharmacological Galectin-3 Inhibition. Sci. Rep. 2019, 9, 9607.
- Kolatsi-Joannou, M.; Price, K.L.; Winyard, P.J.; Long, D.A. Modified citrus pectin reduces galectin-3 expression and disease severity in experimental acute kidney injury. PLoS ONE 2011, 6, e18683.
- Calvier, L.; Martinez-Martinez, E.; Miana, M.; Cachofeiro, V.; Rousseau, E.; Sádaba, J.R.; Zannad, F.; Rossignol, P.; López-Andrés, N. The impact of galectin-3 inhibition on aldosterone-induced cardiac and renal injuries. JACC Heart Fail. 2015, 3, 59–67.
- Martinez-Martinez, E.; Ibarrola, J.; Calvier, L.; Fernandez-Celis, A.; Leroy, C.; Cachofeiro, V.; Rossignol, P.; Lopez-Andres, N. Galectin-3 blockade reduces renal fibrosis in two normotensive experimental models of renal damage. PLoS ONE 2016, 11, e0166272.
- Martínez-Martínez, E.; Ibarrola, J.; Fernández-Celis, A.; Calvier, L.; Leroy, C.; Cachofeiro, V.; Rossignol, P.; López-Andrés, N. Galectin-3 pharmacological inhibition attenuates early renal damage in spontaneously hypertensive rats. J. Hypertens. 2018, 36, 368–376.
- Martinez-Martinez, E.; Calvier, L.; Rossignol, P.; Rousseau, E.; Fernandez-Celis, A.; Jurado-Lopez, R.; Laville, M.; Cachofeiro, V.; Lopez-Andres, N. Galectin-3 inhibition prevents adipose tissue remodelling in obesity. Int. J. Obes. (Lond.) 2016, 40, 1034–1038.
- Abu-Elsaad, N.M.; Elkashef, W.F. Modified citrus pectin stops progression of liver fibrosis by inhibiting galectin-3 and inducing apoptosis of stellate cells. Can. J. Physiol. Pharm. 2016, 94, 554–562.
- Eliaz, I.; Hotchkiss, A.T.; Fishman, M.L.; Rode, D. The effect of modified citrus pectin on urinary excretion of toxic elements. Phytother. Res. 2006, 20, 859–864.
- Eliaz, I.; Weil, E.; Wilk, B. Integrative medicine and the role of modified citrus pectin/alginates in heavy metal chelation and detoxification-five case reports. Komplementmed 2007, 14, 358–364.
- Zhao, Z.Y.; Liang, L.; Fan, X.; Yu, Z.; Hotchkiss, A.T.; Wilk, B.J.; Eliaz, I. The role of modified citrus pectin as an effective chelator of lead in children hospitalized with toxic lead levels. Altern. Health Med. 2008, 14, 34–38.
- Eliaz, I.; Weil, E.; Schwarzbach, J.; Wilk, B. Modified Citrus Pectin/Alginate Dietary Supplement Increased Fecal Excretion of Uranium: A Family. Altern. Health Med. 2019, 25, 20–24.
- Ramachandran, C.; Wilk, B.J.; Hotchkiss, A.; Chau, H.; Eliaz, I.; Melnick, S.J. Activation of human T-helper/inducer cell, T-cytotoxic cell, B-cell, and natural killer (NK)-cells and induction of natural killer cell activity against K562 chronic myeloid leukemia cells with modified citrus pectin. BMC Complement. Altern. Med. 2011, 11, 59.
- Dahdouh, E.; El-Khatib, S.; Baydoun, E.; Abdel-Massih, R.M. Additive Effect of MCP in Combination with Cefotaxime Against Staphylococcus aureus. Med. Chem. 2017, 13, 682–688.
- Ramachandran, C.; Wilk, B.; Melnick, S.J.; Eliaz, I. Synergistic antioxidant and anti-inflammatory effects between modified citrus pectin and honokiol. Evid. Based Complement. Altern. Med. 2017, 2017, 8379843.
- Di, R.; Vakkalanka, M.S.; Onumpai, C.; Chau, H.K.; White, A.; Rastall, R.A.; Yam, K.; Hotchkiss, A.T., Jr. Pectic oligosaccharide structure-function relationships: Prebiotics, inhibitors of Escherichia coli O157: H7 adhesion and reduction of Shiga toxin cytotoxicity in HT29 cells. Food Chem. 2017, 227, 245–254.
- Odun-Ayo, F.; Mellem, J.; Reddy, L. The effect of modified citrus pectin-probiotic on faecal lactobacilli in Balb/c mice. Food Sci. Technol. 2017, 37, 478–482.
- Merheb, R.; Abdel-Massih, R.M.; Karam, M.C. Immunomodulatory effect of natural and modified Citrus pectin on cytokine levels in the spleen of BALB/c mice. Int. J. Biol. Macromol. 2019, 121, 1–5.
- Leclere, L.; Fransolet, M.; Cote, F.; Cambier, P.; Arnould, T.; Van Cutsem, P.; Michiels, C. Heat-modified citrus pectin induces apoptosis-like cell death and autophagy in HepG2 and A549 cancer cells. PLoS ONE 2015, 10, e0115831.
- MacKinnon, A.C.; Liu, X.; Hadoke, P.W.; Miller, M.R.; Newby, D.E.; Sethi, T. Inhibition of galectin-3 reduces atherosclerosis in apolipoprotein E-deficient mice. Glycobiology 2013, 23, 654–663.
- Xu, Y.; Gu, X.; Gong, M.; Guo, G.; Han, K.; An, R. Galectin-3 inhibition sensitizes human renal cell carcinoma cells to arsenic trioxide treatment. Cancer Biol. 2013, 14, 897–906.
- Menachem, A.; Bodner, O.; Pastor, J.; Raz, A.; Kloog, Y. Inhibition of malignant thyroid carcinoma cell proliferation by Ras and galectin-3 inhibitors. Cell Death Discov. 2015, 1, 15047.
- Ruvolo, P.P.; Ruvolo, V.R.; Benton, C.B.; AlRawi, A.; Burks, J.K.; Schober, W.; Rolke, J.; Tidmarsh, G.; Hail, N., Jr.; Davis, R.E. Combination of galectin inhibitor GCS-100 and BH3 mimetics eliminates both p53 wild type and p53 null AML cells. Biochim. Biophys. Acta 2016, 1863, 562–571.
- Clark, M.C.; Pang, M.; Hsu, D.K.; Liu, F.-T.; De Vos, S.; Gascoyne, R.D.; Said, J.; Baum, L.G. Galectin-3 binds to CD45 on diffuse large B-cell lymphoma cells to regulate susceptibility to cell death. Blood 2012, 120, 4635–4644.
- Demotte, N.; Wieërs, G.; Van Der Smissen, P.; Moser, M.; Schmidt, C.; Thielemans, K.; Squifflet, J.-L.; Weynand, B.; Carrasco, J.; Lurquin, C. A galectin-3 ligand corrects the impaired function of human CD4 and CD8 tumor-infiltrating lymphocytes and favors tumor rejection in mice. Cancer Res. 2010, 70, 7476–7488.
- Streetly, M.J.; Maharaj, L.; Joel, S.; Schey, S.A.; Gribben, J.G.; Cotter, F.E. GCS-100, a novel galectin-3 antagonist, modulates MCL-1, NOXA, and cell cycle to induce myeloma cell death. Blood 2010, 115, 3939–3948.
- Wang, Y.; Nangia-Makker, P.; Balan, V.; Hogan, V.; Raz, A. Calpain activation through galectin-3 inhibition sensitizes prostate cancer cells to cisplatin treatment. Cell Death Dis. 2010, 1, e101.
- Volarevic, V.; Markovic, B.S.; Bojic, S.; Stojanovic, M.; Nilsson, U.; Leffler, H.; Besra, G.S.; Arsenijevic, N.; Paunovic, V.; Trajkovic, V. Gal-3 regulates the capacity of dendritic cells to promote NKT-cell-induced liver injury. Eur. J. Immunol. 2015, 45, 531–543.
- Volarevic, V.; Milovanovic, M.; Ljujic, B.; Pejnovic, N.; Arsenijevic, N.; Nilsson, U.; Leffler, H.; Lukic, M.L. Galectin-3 deficiency prevents concanavalin A–induced hepatitis in mice. Hepatology 2012, 55, 1954–1964.
- MacKinnon, A.C.; Gibbons, M.A.; Farnworth, S.L.; Leffler, H.; Nilsson, U.J.; Delaine, T.; Simpson, A.J.; Forbes, S.J.; Hirani, N.; Gauldie, J. Regulation of transforming growth factor-β1–driven lung fibrosis by galectin-3. Am. J. Respir. Crit. Care Med. 2012, 185, 537–546.
- Traber, P.G.; Chou, H.; Zomer, E.; Hong, F.; Klyosov, A.; Fiel, M.-I.; Friedman, S.L. Regression of fibrosis and reversal of cirrhosis in rats by galectin inhibitors in thioacetamide-induced liver disease. PLoS ONE 2013, 8, e75361.
- Traber, P.G.; Zomer, E. Therapy of experimental NASH and fibrosis with galectin inhibitors. PLoS ONE 2013, 8, e83481.
More