In the search for new therapeutic strategies to contrast SARS-CoV-2, we here studied the interaction of polydatin (PD) and resveratrol (RESV)—two natural stilbene polyphenols with manifold, well known biological activities—with Spike, the viral protein essential for virus entry into host cells, and ACE2, the angiotensin-converting enzyme present on the surface of multiple cell types (including respiratory epithelial cells) which is the main host receptor for Spike binding. Molecular Docking simulations evidenced that both compounds can bind Spike, ACE2 and the ACE2:Spike complex with good affinity, although the interaction of PD appears stronger than that of RESV on all the investigated targets. Preliminary biochemical assays revealed a significant inhibitory activity of the ACE2:Spike recognition with a dose-response effect only in the case of PD.
- SARS-CoV-2
- polydatin
- resveratrol
- molecular docking
- protein-binding
- ACE2:Spike binding-inhibition
1. Introduction
2. Molecular docking simulation.
Spike-protein pre-fusion conformation [50,76] is a trimer constituted of two subunits, S1 and S2, which are cleaved following receptor binding [77]. S1 Receptor Binding Domains (RBDs) host the binding motifs (RBMs) able to recognize ACE2. The high RBD flexibility allows the Spike to sample open or closed conformations, in which RBMs are respectively exposed or hidden inside the protomers interface [77–81].
Therefore, the binding to SARS-CoV-2 Spike structure with one RBD in an open conformation (PDB ID: 6VSB [50]) has been investigated by molecular docking simulations. The pockets on the RBD surface appear able to accommodate both PD and RESV ligands (Figure 2). A slightly lower affinity for the RBD domain was found for RESV (−6.5 kcal/mol for the best docking pose, Table 1) with respect to PD (top-ranked pose −6.9 kcal/mol).
Table 1. Docking scores (i.e., the approximate binding energy estimated by the docking scoring function, kcal/mol units) for PD and RESV top-ranked docked poses.
|
|
PD |
RESV |
|
Spike RBD |
−6.9 |
−6.5 |
|
ACE2 |
−8.4 |
−6.9 |
|
S:ACE2 region I |
−8.1 |
−7.6 |
|
S:ACE2 region II |
−6.9 |
−6.5 |
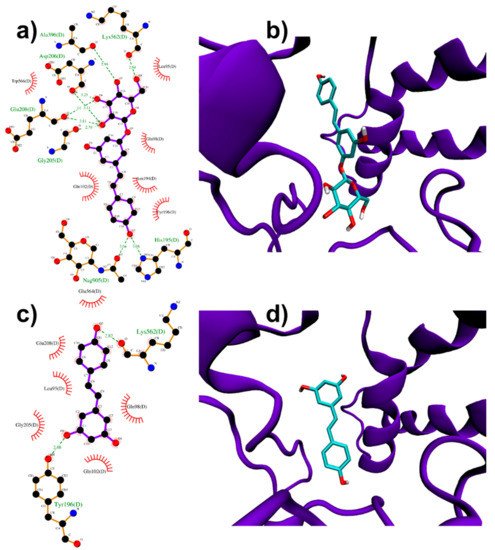
ACE2 homodimer (PDB ID: 6M18, [51]) is constituted by an N-terminal protease domain, involved in the interaction with SARS-CoV-2 Spike RBD [51], and a C-terminal collectrin-like domain, comprising the ACE2 transmembrane helix. Molecular docking simulations were performed near ACE2 protease domain α1 and α2 helices, thus far from the catalytic site related to ACE2 physiological function. Despite its size, PD can be easily accommodated in a deep groove behind the two helices (Figure 3a,b) producing a high docking score (−8.4 kcal/mol, Table 1). RESV can also bind to the pockets near α1 and α2 helices, but with a ~1 kcal/mol lower score with respect to PD (−6.9 kcal/mol, Table 1) (Figure 3c,d).
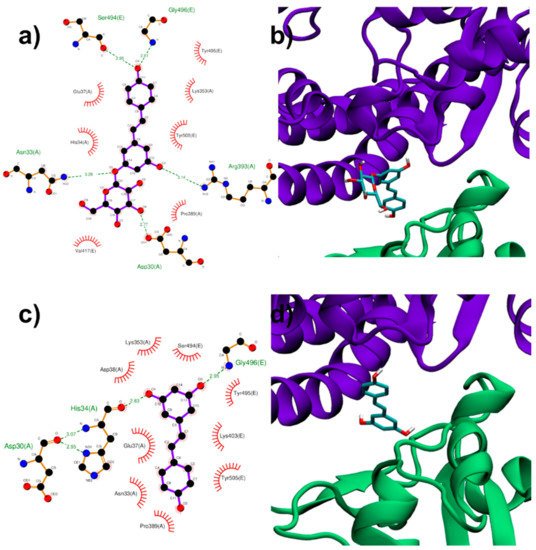
Both RESV and PD showed a good estimated binding affinity in the groove between the Spike RBD and ACE2. Due to the high spatial extent of the Spike:ACE2 interface, docking simulations were performed in two distinct regions (named regions I and II). In the first interfacial region investigated (region I), the most stable RESV binding mode involves recognition of one of the interfacial pockets through polar and hydrophobic contacts (Figure 4c,d). PD binds in the same pocket as RESV (Figure 4a,b) but interacting with more surrounding residues, thus showing a higher docking score (−8.1 kcal/mol, Table 1), compared to that of RESV (−7.6 kcal/mol).
| PD | RESV | |
|---|---|---|
| Spike RBD | −6.9 | −6.5 |
| Spike A/B interface | −7.3 | >−6.5 |
| Spike A/C interface | −7.3 | >−6.5 |
| ACE2 | −8.4 | −6.9 |
| S:ACE2 region I | −8.1 | −7.6 |
| S:ACE2 region II | −6.9 | −6.5 |
In the second region (region II) investigated at the interface between viral Spike RBD and human ACE2, the top-ranked RESV conformer fits into a large ACE2 cavity far from the interaction sites between the two protein domains. On the contrary, in the best PD binding mode, the ligand binds onto a pocket located at one end of the ACE2:Spike complex interface with a docking score 0.4 kcal/mol lower than RESV (−6.9 kcal/mol, Table 1). It is interesting to note the fundamental role played by the glucosidic moiety which interacts with the residues of the main chains that are involved in the formation of the ACE2:Spike dimer. Weakening of these interactions could, in principle, contribute to the dissociation of the dimeric complex.
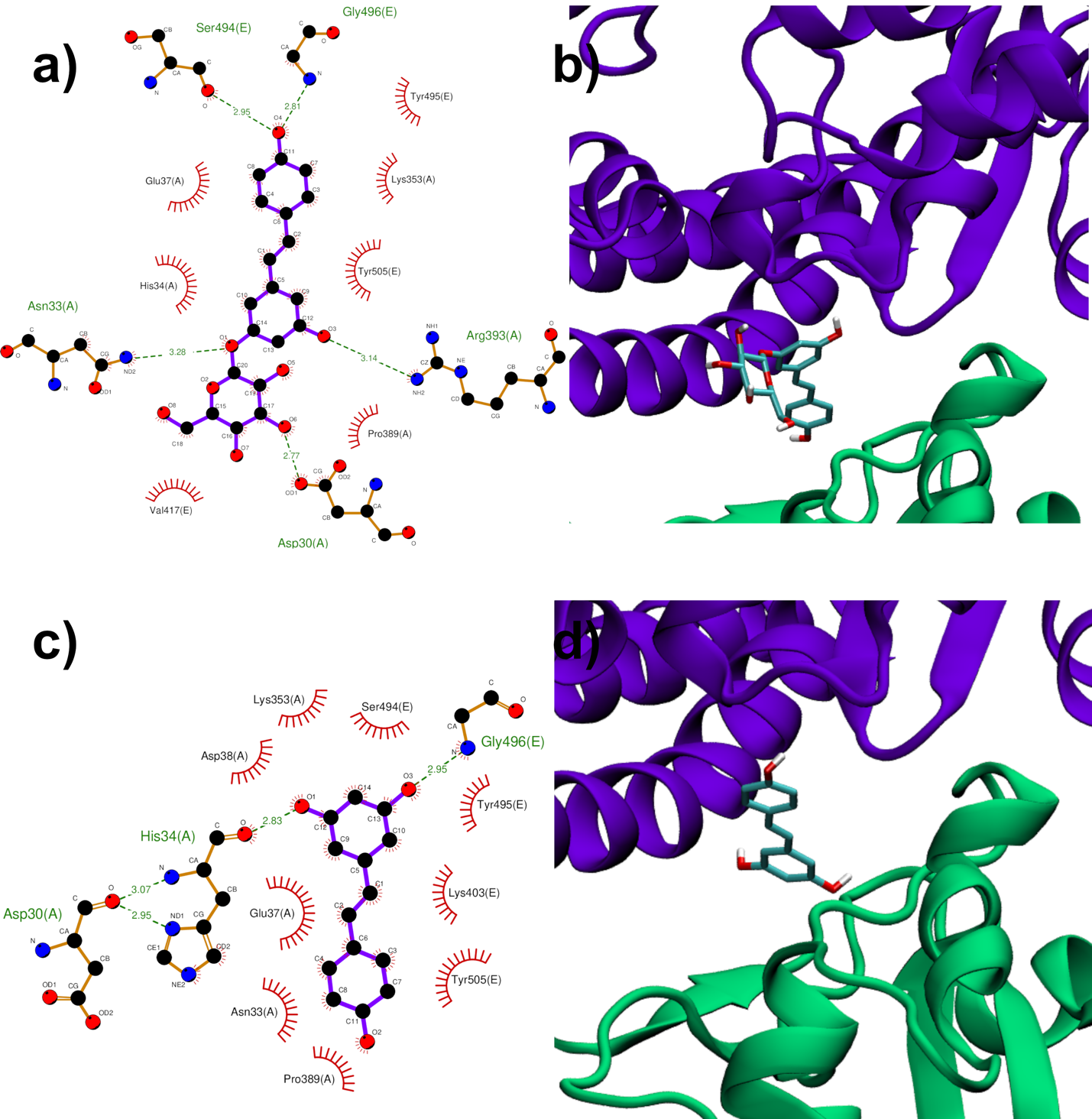
Figure 4. PD (a,b) and RESV (c,d) top-ranked poses docked to Spike:ACE2 interface region I. Two-dimensional interaction maps (a–c): C, N and O atoms are reported in black, blue and red, respectively. Hydrogen bonds are depicted as green dashed lines, while hydrophobic interactions as red cogwheels. Hydrogen atoms are not depicted for ease of illustration. The names of proteins’ residues involved in interactions with the ligand are reported. Three-dimensional representations of PD and RESV docked poses (b–d): protets from dockins’ backbone is represented as a cartoon, while the ligand as sticks.
Results fro sim docking simulations on the already assembled Spike:ACE2 complex reveal the potential capability of PD, but also RESV, to insert themselves into the extended adduct interface. This leads to the hypothesis of a ligand-induced dissociation or weakening effect, through allosteric (such as for region I) or direct (region II) interference.
3. To establish if RESV and PD can experimentally interfere with the binding of the Spike protein with ACE2 receptor, as suggested by the molecular docking simulations, binding inhibition assays were performed.
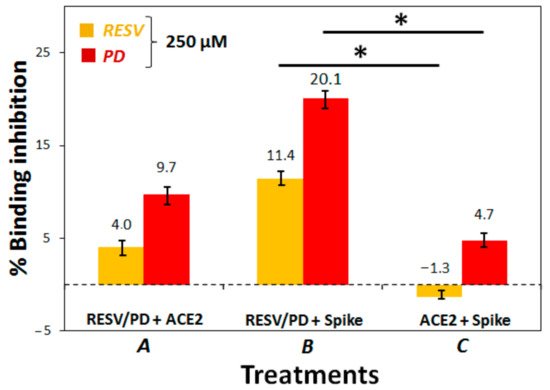
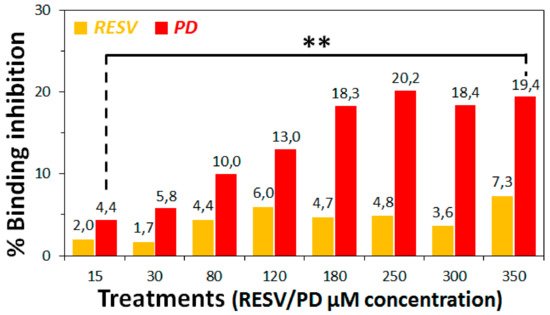
4. Conclusions
References
- Fehr, A.R.; Perlman, S. Coronaviruses: An Overview of Their Replication and Pathogenesis. In Coronaviruses: Methods and Protocols; Springer: New York, NY, USA, 2015; pp. 1–23.
- Masters, P.S. The Molecular Biology of Coronaviruses. Adv. Virus Res. 2006, 65, 193–292.
- Corman, V.M.; Lienau, J.; Witzenrath, M. Coronaviruses as the Cause of Respiratory Infections. Internist 2019, 60, 1136–1145.
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical Features of Patients Infected with 2019 Novel Coronavirus in Wuhan, China. Lancet 2020, 395, 497–506.
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269.
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin. Nature 2020, 579, 270–273.
- Siu, Y.L.; Teoh, K.T.; Lo, J.; Chan, C.M.; Kien, F.; Escriou, N.; Tsao, S.W.; Nicholls, J.M.; Altmeyer, R.; Peiris, J.S.M.; et al. The M, E, and N Structural Proteins of the Severe Acute Respiratory Syndrome Coronavirus Are Required for Efficient Assembly, Trafficking, and Release of Virus-Like Particles. J. Virol. 2008, 82, 11318–11330.
- Belouzard, S.; Millet, J.K.; Licitra, B.N.; Whittaker, G.R. Mechanisms of Coronavirus Cell Entry Mediated by the Viral Spike Protein. Viruses 2012, 4, 1011–1033.
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell Entry Mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734.
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural Basis of Receptor Recognition by SARS-CoV-2. Nature 2020, 581, 221–224.
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, e00127-20.
- Bourgonje, A.R.; Abdulle, A.E.; Timens, W.; Hillebrands, J.L.; Navis, G.J.; Gordijn, S.J.; Bolling, M.C.; Dijkstra, G.; Voors, A.A.; Osterhaus, A.D.M.E.; et al. Angiotensin-Converting Enzyme 2 (ACE2), SARS-CoV-2 and the Pathophysiology of Coronavirus Disease 2019 (COVID-19). J. Pathol. 2020, 251, 228–248.
- Lukassen, S.; Chua, R.L.; Trefzer, T.; Kahn, N.C.; Schneider, M.A.; Muley, T.; Winter, H.; Meister, M.; Veith, C.; Boots, A.W.; et al. SARS-CoV-2 Receptor ACE 2 and TMPRSS 2 Are Primarily Expressed in Bronchial Transient Secretory Cells. EMBO J. 2020, 39, e105114.
- Magrone, T.; Magrone, M.; Jirillo, E. Focus on Receptors for Coronaviruses with Special Reference to Angiotensin- Converting Enzyme 2 as a Potential Drug Target—A Perspective. Endocr. Metab. Immune Disord. Drug Targets 2020, 20, 807–811.
- Chi, X.; Yan, R.; Zhang, J.; Zhang, G.; Zhang, Y.; Hao, M.; Zhang, Z.; Fan, P.; Dong, Y.; Yang, Y.; et al. A Neutralizing Human Antibody Binds to the N-Terminal Domain of the Spike Protein of SARS-CoV-2. Science 2020, 369, 650–655.
- Liu, L.; Wang, P.; Nair, M.S.; Yu, J.; Rapp, M.; Wang, Q.; Luo, Y.; Chan, J.F.W.; Sahi, V.; Figueroa, A.; et al. Potent Neutralizing Antibodies against Multiple Epitopes on SARS-CoV-2 Spike. Nature 2020, 584, 450–456.
- Papageorgiou, A.C.; Mohsin, I. The SARS-CoV-2 Spike Glycoprotein as a Drug and Vaccine Target: Structural Insights into Its Complexes with ACE2 and Antibodies. Cells 2020, 9, 2343.
- Yi, C.; Sun, X.; Ye, J.; Ding, L.; Liu, M.; Yang, Z.; Lu, X.; Zhang, Y.; Ma, L.; Gu, W.; et al. Key Residues of the Receptor Binding Motif in the Spike Protein of SARS-CoV-2 That Interact with ACE2 and Neutralizing Antibodies. Cell. Mol. Immunol. 2020, 17, 621–630.
- Ju, B.; Zhang, Q.; Ge, J.; Wang, R.; Sun, J.; Ge, X.; Yu, J.; Shan, S.; Zhou, B.; Song, S.; et al. Human Neutralizing Antibodies Elicited by SARS-CoV-2 Infection. Nature 2020, 584, 115–119.
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8.
- Gil, C.; Ginex, T.; Maestro, I.; Nozal, V.; Barrado-Gil, L.; Cuesta-Geijo, M.Á.; Urquiza, J.; Ramírez, D.; Alonso, C.; Campillo, N.E.; et al. COVID-19: Drug Targets and Potential Treatments. J. Med. Chem. 2020, 63, 12359–12386.
- Hensel, A.; Bauer, R.; Heinrich, M.; Spiegler, V.; Kayser, O.; Hempel, G.; Kraft, K. Challenges at the Time of COVID-19: Opportunities and Innovations in Antivirals from Nature. Planta Med. 2020, 86, 659–664.
- Islam, M.T.; Sarkar, C.; El-Kersh, D.M.; Jamaddar, S.; Uddin, S.J.; Shilpi, J.A.; Mubarak, M.S. Natural Products and Their Derivatives against Coronavirus: A Review of the Non-clinical and Pre-clinical Data. Phyther. Res. 2020, 34, 2471–2492.
- Mani, J.S.; Johnson, J.B.; Steel, J.C.; Broszczak, D.A.; Neilsen, P.M.; Walsh, K.B.; Naiker, M. Natural Product-Derived Phytochemicals as Potential Agents against Coronaviruses: A Review. Virus Res. 2020, 284, 197989.
- Romeo, I.; Mesiti, F.; Lupia, A.; Alcaro, S. Current Updates on Naturally Occurring Compounds Recognizing SARS-CoV-2 Druggable Targets. Molecules 2021, 26, 632.
- Quideau, S.; Deffieux, D.; Douat-Casassus, C.; Pouységu, L. Plant Polyphenols: Chemical Properties, Biological Activities, and Synthesis. Angew. Chemie Int. Ed. 2011, 50, 586–621.
- Gambini, J.; Inglés, M.; Olaso, G.; Lopez-Grueso, R.; Bonet-Costa, V.; Gimeno-Mallench, L.; Mas-Bargues, C.; Abdelaziz, K.M.; Gomez-Cabrera, M.C.; Vina, J.; et al. Properties of Resveratrol: In Vitro and In Vivo Studies about Metabolism, Bioavailability, and Biological Effects in Animal Models and Humans. Oxid. Med. Cell. Longev. 2015, 2015, 837042.
- Du, Q.H.; Peng, C.; Zhang, H. Polydatin: A Review of Pharmacology and Pharmacokinetics. Pharm. Biol. 2013, 51, 1347–1354.
- Zorzete, P.; Reis, T.A.; Felício, J.D.; Baquião, A.C.; Makimoto, P.; Corrêa, B. Fungi, Mycotoxins and Phytoalexin in Peanut Varieties, during Plant Growth in the Field. Food Chem. 2011, 129, 957–964.
- Regev-Shoshani, G.; Shoseyov, O.; Bilkis, I.; Kerem, Z. Glycosylation of Resveratrol Protects It from Enzymic Oxidation. Biochem. J. 2003, 374, 157–163.
- Fabris, S.; Momo, F.; Ravagnan, G.; Stevanato, R. Antioxidant Properties of Resveratrol and Piceid on Lipid Peroxidation in Micelles and Monolamellar Liposomes. Biophys. Chem. 2008, 135, 76–83.
- Platella, C.; Guida, S.; Bonmassar, L.; Aquino, A.; Bonmassar, E.; Ravagnan, G.; Montesarchio, D.; Roviello, G.N.; Musumeci, D.; Fuggetta, M.P. Antitumour Activity of Resveratrol on Human Melanoma Cells: A Possible Mechanism Related to Its Interaction with Malignant Cell Telomerase. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2843–2851.
- Platella, C.; Raucci, U.; Rega, N.; D’Atri, S.; Levati, L.; Roviello, G.N.; Fuggetta, M.P.; Musumeci, D.; Montesarchio, D. Shedding Light on the Interaction of Polydatin and Resveratrol with G-Quadruplex and Duplex DNA: A Biophysical, Computational and Biological Approach. Int. J. Biol. Macromol. 2020, 151, 1163–1172.
- Francioso, A.; Mastromarino, P.; Masci, A.; D’Erme, M.; Mosca, L. Chemistry, Stability and Bioavailability of Resveratrol. Med. Chem. 2014, 10, 237–245.
- Şöhretoğlu, D.; Baran, M.Y.; Arroo, R.; Kuruüzüm-Uz, A. Recent Advances in Chemistry, Therapeutic Properties and Sources of Polydatin. Phytochem. Rev. 2018, 17, 973–1005.
- Magrone, T.; Magrone, M.; Russo, M.A.; Jirillo, E. Recent Advances on the Anti-Inflammatory and Antioxidant Properties of Red Grape Polyphenols: In Vitro and In Vivo Studies. Antioxidants 2019, 9, 35.
- Dong, L.; Hu, S.; Gao, J. Discovering Drugs to Treat Coronavirus Disease 2019 (COVID-19). Drug Discov. Ther. 2020, 14, 58–60.
- Wahedi, H.M.; Ahmad, S.; Abbasi, S.W. Stilbene-Based Natural Compounds as Promising Drug Candidates against COVID-19. J. Biomol. Struct. Dyn. 2020, 39, 3225–3234.
- Yang, M.; Wei, J.; Huang, T.; Lei, L.; Shen, C.; Lai, J.; Yang, M.; Liu, L.; Yang, Y.; Liu, G.; et al. Resveratrol Inhibits the Replication of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) in Cultured Vero Cells. Phyther. Res. 2020, 35, 1127–1129.
- Feng, H.; Choi, H.Y.; Cui, C.; Xu, H.; Jiang, J.; Yan, G.; Jin, M. Effects of Polydatin on Oleic Acid-Induced Acute Respiratory Distress Syndrome in Rats. Int. J. Clin. Exp. Med. 2018, 11, 12106–12114.
- Filardo, S.; Di Pietro, M.; Mastromarino, P.; Sessa, R. Therapeutic Potential of Resveratrol against Emerging Respiratory Viral Infections. Pharmacol. Ther. 2020, 214, 107613.
- Jiang, Q.; Yi, M.; Guo, Q.; Wang, C.; Wang, H.; Meng, S.; Liu, C.; Fu, Y.; Ji, H.; Chen, T. Protective Effects of Polydatin on Lipopolysaccharide-Induced Acute Lung Injury through TLR4-MyD88-NF-ΚB Pathway. Int. Immunopharmacol. 2015, 29, 370–376.
- Khalil, A.; Tazeddinova, D. The Upshot of Polyphenolic Compounds on Immunity amid COVID-19 Pandemic and Other Emerging Communicable Diseases: An Appraisal. Nat. Products Bioprospect. 2020, 10, 411–429.
- Li, X.H.; Gong, X.; Zhang, L.; Jiang, R.; Li, H.Z.; Wu, M.J.; Wan, J.Y. Protective Effects of Polydatin on Septic Lung Injury in Mice via Upregulation of HO-1. Mediators Inflamm. 2013, 2013, 1–10.
- Lanzilli, G.; Cottarelli, A.; Nicotera, G.; Guida, S.; Ravagnan, G.; Fuggetta, M.P. Anti-Inflammatory Effect of Resveratrol and Polydatin by in Vitro IL-17 Modulation. Inflammation 2012, 35, 240–248.
- Lo Muzio, L.; Bizzoca, M.E.; Ravagnan, G. New Intriguing Possibility for Prevention of Coronavirus Pneumonitis: Natural Purified Polyphenols. Oral Dis. 2020, odi.13518.
- Yan, X.-D.; Wang, Q.-M.; Tie, C.; Jin, H.-T.; Han, Y.-X.; Zhang, J.-L.; Yu, X.-M.; Hou, Q.; Zhang, P.-P.; Wang, A.-P.; et al. Polydatin Protects the Respiratory System from PM2.5 Exposure. Sci. Rep. 2017, 7, 40030.
- Ravagnan, G.; De Filippis, A.; Cartenì, M.; De Maria, S.; Cozza, V.; Petrazzuolo, M.; Tufano, M.A.; Donnarumma, G. Polydatin, A Natural Precursor of Resveratrol, Induces β-Defensin Production and Reduces Inflammatory Response. Inflammation 2013, 36, 26–34.
- Pasquereau, S.; Nehme, Z.; Haidar Ahmad, S.; Daouad, F.; Van Assche, J.; Wallet, C.; Schwartz, C.; Rohr, O.; Morot-Bizot, S.; Herbein, G. Resveratrol Inhibits HCoV-229E and SARS-CoV-2 Coronavirus Replication In Vitro. Viruses 2021, 13, 354.
- López-Nicolás, J.M.; Pérez-Gilabert, M.; García-Carmona, F. Effect of Protonation and Aggregation State of (E)-Resveratrol on Its Hydroperoxidation by Lipoxygenase. J. Agric. Food Chem. 2009, 57, 4630–4635.
- López-Nicolás, J.M.; García-Carmona, F. Aggregation State and PKa Values of (E)-Resveratrol As Determined by Fluorescence Spectroscopy and UV−Visible Absorption. J. Agric. Food Chem. 2008, 56, 7600–7605.
- Polidase®, the Bioavailable Resveratrol, Sherman Tree Nutraceuticals. Available online: https://shermantree.it/en/polidase/ (accessed on 9 July 2021).
- Izzo, G.M.; Suffritti, G. Polydatin and Atopic Dermatitis in Adults: Clinical Study. J. Cosmetol. Trichol. 2017, 3, 122.
- Cremon, C.; Stanghellini, V.; Barbaro, M.R.; Cogliandro, R.F.; Bellacosa, L.; Santos, J.; Vicario, M.; Pigrau, M.; Alonso Cotoner, C.; Lobo, B.; et al. Randomised Clinical Trial: The Analgesic Properties of Dietary Supplementation with Palmitoylethanolamide and Polydatin in Irritable Bowel Syndrome. Aliment. Pharmacol. Ther. 2017, 45, 909–922.
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural Basis for the Recognition of SARS-CoV-2 by Full-Length Human ACE2. Science 2020, 367, 1444–1448.
- Bonucci, M.; Raggi, R.; Vacca, R.A. Polydatin and Its Potential Protective Effect on COVID-19. Clin. Nutr. 2020, 39, 3850–3851.