Regulated cell death (RCD) is a fundamental process common to nearly all living beings and essential for the development and tissue homeostasis in animals and humans. A wide range of molecules can induce RCD including a number of viral proteolytic enzymes. To date, numerous data indicate that picornaviral 3C proteases can induce RCD. In most reported cases, these proteases induce classical caspase-dependent apoptosis. In contrast, the human hepatitis A virus 3C protease (3Cpro) has recently been shown to cause caspase-independent cell death accompanied by previously undescribed features.
In the current topic the results of the study where 3Cpro-induced cell death was characterized morphologically and biochemically are presented. It was found that dead cells demonstrated necrosis-like morphological changes including permeabilization of plasma membrane, loss of mitochondrial potential, as well as mitochondria and nuclei swelling. Additionally, it was shown that 3Cpro-induced cell death was efficiently blocked by ferroptosis inhibitors and was accompanied by intense lipid peroxidation. Taken together, these results indicate that 3Cpro induces ferroptosis upon its individual expression in human cells. This is the first demonstration that a proteolytic enzyme can induce ferroptosis, the recently discovered and actively studied type of RCD.
- hepatitis A virus
- 3C protease
- regulated cell death
- ferroptosis
- ectopic expression
1. Introduction
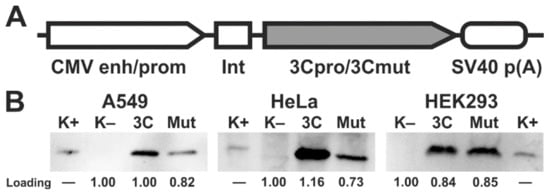
2. Ectopic Expression of 3Cpro and Its Inactive Form 3Cmut in Human Cells
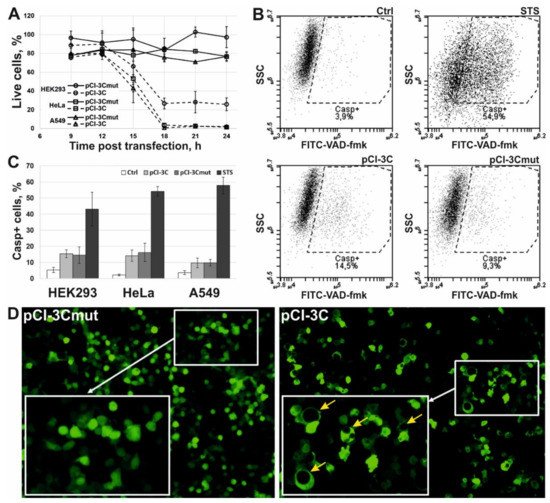
3. 3Cpro Expression Induces Caspase-Independent Cell Death with Cytoplasmic Vacuolization
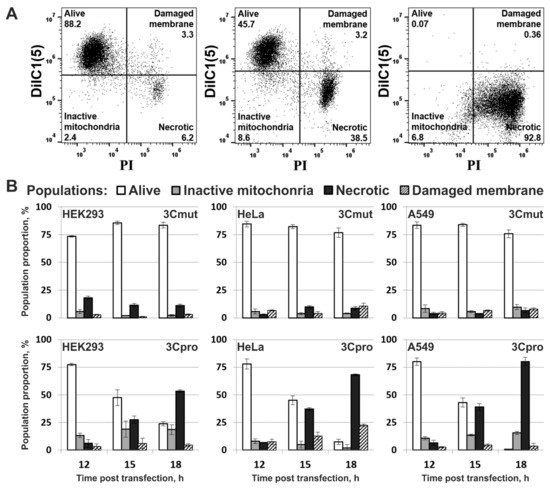
4. Cells Expressing 3Cpro Acquire Necrotic Morphology and Are Characterized by Nuclei and Mitochondria Swelling
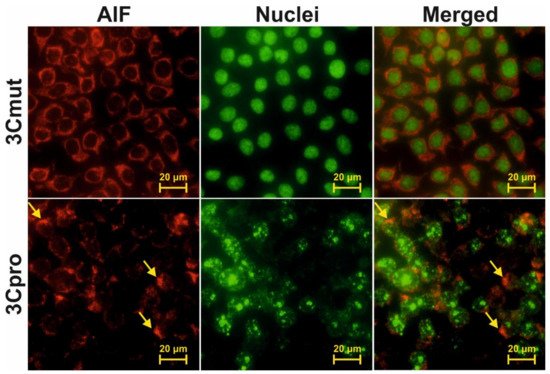
5. 3Cpro-Induced Cell Death Is Effectively Blocked by Ferroptosis Inhibitors and Is Accompinied by Lipid Peroxidation
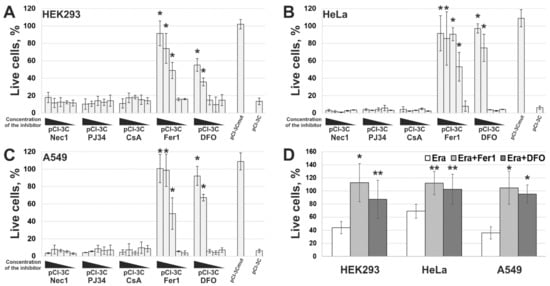
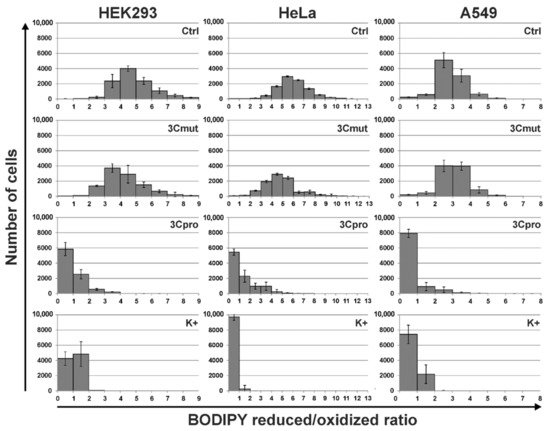
However, the data obtained are not enough to establish the biological role of 3Cpro as a ferroptosis inducer. It is common knowledge that human hepatitis A virus has no direct cytopathic effect on hepatocytes, although the liver is the primary locus of virus replication. In vivo the main factor that damages the liver is the death of infected hepatocytes mainly due to the activity of cytotoxic T cells and natural killer cells [29][30]. Moreover, the intracellular level of 3Cpro is apparently much lower during the infection compared to that in our experimental system. In this context, it is likely that 3Cpro affects certain cell substrates to maintain viral replication in vivo, while ferroptosis induction is a side effect of 3Cpro action. Apparently, low cellular levels of 3Cpro have no such side effect, while higher protease levels in our experimental system can induce it. A detailed analysis of the molecular mechanism of 3Cpro-induced cell death is needed to reveal the relationship between the ability of 3Cpro to induce ferroptosis and the viral life cycle. In the first place, the cellular targets of 3Cpro should be identified. This information can also extend our knowledge about the mechanism and biological role of ferroptosis.
References
- Allocati, N.; Masulli, M.; Di Ilio, C.; De Laurenzi, V. Die for the community: An overview of programmed cell death in bacteria. Cell Death Dis. 2015, 6, e1609.
- Fuchs, Y.; Steller, H. Programmed Cell Death in Animal Development and Disease. Cell 2011, 147, 742–758.
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541.
- Laitinen, O.; Svedin, E.; Kapell, S.; Nurminen, A.; Hytönen, V.; Flodström-Tullberg, M. Enteroviral proteases: Structure, host interactions and pathogenicity. Rev. Med. Virol. 2016, 26, 251–267.
- Sun, D.; Chen, S.; Cheng, A.; Wang, M. Roles of the Picornaviral 3C Proteinase in the Viral Life Cycle and Host Cells. Viruses 2016, 8, 82.
- Calandria, C.; Irurzun, A.; Barco, A.; Carrasco, L. Individual expression of poliovirus 2Apro and 3Cpro induces activation of caspase-3 and PARP cleavage in HeLa cells. Virus Res. 2004, 104, 39–49.
- Li, M.-L.; Hsu, T.-A.; Chen, T.-C.; Chang, S.-C.; Lee, J.-C.; Chen, C.-C.; Stollar, V.; Shih, S.-R. The 3C Protease Activity of Enterovirus 71 Induces Human Neural Cell Apoptosis. Virology 2002, 293, 386–395.
- Chau, D.H.W.; Yuan, J.; Zhang, H.; Cheung, P.; Lim, T.; Liu, Z.; Sall, A.; Yang, D. Coxsackievirus B3 proteases 2A and 3C induce apoptotic cell death through mitochondrial injury and cleavage of eIF4GI but not DAP5/p97/NAT1. Apoptosis 2006, 12, 513–524.
- Lötzerich, M.; Roulin, P.S.; Boucke, K.; Witte, R.; Georgiev, O.; Greber, U.F. Rhinovirus 3C protease suppresses apoptosis and triggers caspase-independent cell death. Cell Death Dis. 2018, 9, 1–18.
- Shubin, A.V.; Lunina, N.A.; Shedova, E.N.; Roshina, M.P.; Demidyuk, I.V.; Vinogradova, T.V.; Kopantsev, E.P.; Chernov, I.P.; Kostrov, S.V. Evaluation of the toxic effects evoked by the transient expression of protease genes from human pathogens in HEK293 cells. Appl. Biochem. Microbiol. 2013, 49, 750–755.
- Shubin, A.V.; Demidyuk, I.V.; Lunina, N.A.; Komissarov, A.A.; Roschina, M.P.; Leonova, O.G.; Kostrov, S.V. Protease 3C of hepatitis A virus induces vacuolization of lysosomal/endosomal organelles and caspase-independent cell death. BMC Cell Biol. 2015, 16, 4.
- Shubin, A.V.; Komissarov, A.A.; Karaseva, M.A.; Padman, B.S.; Kostrov, S.V.; Demidyuk, I.V. Human hepatitis A virus 3C protease exerts a cytostatic effect on Saccharomyces cerevisiae and affects the vacuolar compartment. Biologia 2021, 76, 321–327.
- Komissarov, A.; Karaseva, M.A.; Safina, D.R.; Roschina, M.P.; Bednova, O.P.; Kazakov, A.A.; Demkin, V.V.; Demidyuk, I.V. Comparative evaluation of the transgene expression efficiency provided by the model genetic constructs of different structure. Mol. Genet. Microbiol. Virol. 2016, 31, 156–162.
- Komissarov, A.; Demidyuk, I.; Safina, D.; Roschina, M.; Shubin, A.; Lunina, N.; Karaseva, M.; Kostrov, S. Cytotoxic effect of co-expression of human hepatitis A virus 3C protease and bifunctional suicide protein FCU1 genes in a bicistronic vector. Mol. Biol. Rep. 2017, 44, 323–332.
- Western Blot Normalization Using Image Lab Software, Quick Start Guide. Bio-Rad Bulletin 6434; Bio-Rad Laboratories: Hercules, CA, USA, 2015.
- Bertrand, R.; Solary, E.; O’Connor, P.; Kohn, K.W.; Pommier, Y. Induction of a Common Pathway of Apoptosis by Staurosporine. Exp. Cell Res. 1994, 211, 314–321.
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119.
- Jagtap, P.; Soriano, F.; Virág, L.; Liaudet, L.; Mabley, J.; Szabó, É.; Haskó, G.; Marton, A.; Lorigados, C.B.; Gallyas, F.; et al. Novel phenanthridinone inhibitors of poly(adenosine 5′-diphosphate-ribose) synthetase: Potent cytoprotective and antishock agents. Crit. Care Med. 2002, 30, 1071–1082.
- Kajitani, K.; Fujihashi, M.; Kobayashi, Y.; Shimizu, S.; Tsujimoto, Y.; Miki, K. Crystal structure of human cyclophilin D in complex with its inhibitor, cyclosporin A at 0.96-Å resolution. Proteins Struct. Funct. Bioinform. 2007, 70, 1635–1639.
- Zilka, O.; Shah, R.; Li, B.; Angeli, J.P.F.; Griesser, M.; Conrad, M.; Pratt, D.A. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent. Sci. 2017, 3, 232–243.
- Angeli, J.P.F.; Shah, R.; Pratt, D.A.; Conrad, M. Ferroptosis Inhibition: Mechanisms and Opportunities. Trends Pharmacol. Sci. 2017, 38, 489–498.
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072.
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296.
- Yang, W.S.; Stockwell, B.R. Synthetic Lethal Screening Identifies Compounds Activating Iron-Dependent, Nonapoptotic Cell Death in Oncogenic-RAS-Harboring Cancer Cells. Chem. Biol. 2008, 15, 234–245.
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125.
- Feng, H.; Stockwell, B.R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018, 16, e2006203.
- Lu, B.; Chen, X.B.; Ying, M.D.; He, Q.J.; Cao, J.; Yang, B. The Role of Ferroptosis in Cancer Development and Treatment Response. Front. Pharmacol. 2018, 8, 992.
- Shen, Z.; Song, J.; Yung, B.C.; Zhou, Z.; Wu, A.; Chen, X. Emerging Strategies of Cancer Therapy Based on Ferroptosis. Adv. Mater. 2018, 30, e1704007.
- Fleischer, B.; Fleischer, S.; Maier, K.; Wiedmann, K.H.; Sacher, M.; Thaler, H.; Vallbracht, A. Clonal analysis of infiltrating T lymphocytes in liver tissue in viral hepatitis A. Immunology 1990, 69, 14–19.
- Baba, M.; Hasegawa, H.; Nakayabu, M.; Fukai, K.; Suzuki, S. Cytolytic activity of natural killer cells and lymphokine activated killer cells against hepatitis A virus infected fibroblasts. J. Clin. Lab. Immunol. 1993, 40, 47–60.