Covalent organic frameworks (COFs) are 2D or 3D low density crystalline porous materials with periodically ordered skeletons constituted by organic molecules linked through covalent bonds. They were first reported by Yaghi and collaborators in 2005 from condensation of benzenediboronic acid (BDBA) alone and in the presence of hexahydroxytriphenylene (HHTP) in a simple one-pot procedure at 120 °C, obtaining a boroxine COF (COF-1) and a boronate ester COF (COF-5), respectively. Since then, there has been steady growth in the number of published works dealing with the synthesis, properties, and catalytic applications of COFs.
1. Introduction
Covalent organic frameworks (COFs) are 2D or 3D low density crystalline porous materials with periodically ordered skeletons constituted by organic molecules linked through covalent bonds. They were first reported by Yaghi and collaborators in 2005 from condensation of benzenediboronic acid (BDBA) alone and in the presence of hexahydroxytriphenylene (HHTP) in a simple one-pot procedure at 120 °C, obtaining a boroxine COF (COF-1) and a boronate ester COF (COF-5), respectively [1]. Since then, there has been steady growth in the number of published works dealing with the synthesis, properties, and catalytic applications of COFs. In contrast to other porous organic polymers (POPs), COFs are crystalline (long-range ordered) by definition. In fact, some authors consider POPs to be amorphous COFs, and classify COFs according to their type of long-range order (0D, 1D, 2D, and 3D COFs) [2]. The crystallinity of COFs is governed by their predesigned tailor-made structures, which involve a geometric combination of rigid monomers to guide the growth of polygonal structures [3]. On the other hand, the porosity of COFs is intrinsically linked to the geometry and topicity of the nodes and linkers selected. In fact, one can find COFs with regular pores of a single geometry (trigonal, tetragonal, rhombohedral, or hexagonal), with a mixture of two geometries (two different trigonal ones or hexagonal plus trigonal), or with irregular geometrical pores [3]. The formation of the pores is generally driven by intermolecular interactions, mainly π-stacking and hydrogen bonds [4], but new strategies using templates are being developed [5].
Semiconductors are materials able to harvest light that can be used to induce photochemical transformations, such as the hydrogen evolution reaction (HER), if they exhibit appropriate bandgaps. Under these conditions, electrons are excited from the valence band (VB) to the conduction band (CB) by photons with an energy greater than the bandgap, which are then used to reduce protons to H 2. Historically, semiconductors used for HER photocatalysis have been CdS [6] and Ag 3PO 4 [7], absorbing in the visible region but containing expensive and polluting metals, or graphitic C 3N 4 (g-C 3N 4), which absorbs in the visible range although its bandgap tuning is constrained by its fixed structure [8]. In 2014, Lotsch et al. described for the first time the photocatalytic HER ability of a Pt-doped hydrazone-linked COF derived from 1,3,5-tris-(4-formyl-phenyl)triazine (TFPT-COF) [9]. Thanks to the visible-light-absorbing properties of the crystalline honeycomb-type planar organic scaffold, TFPT-COF can act as the photosensitizer, while Pt acts as the co-catalyst. Afterwards, a large amount of work has been done in this area because COFs show several advantages compared with commonly used semiconductors: (1) they are 100% organic, reducing the price of the photocatalyst; (2) they allow for the tuning of the bandgap by modifying the building blocks; (3) they are mostly obtained under mild reaction conditions (in contrast to g-C 3N 4); (4) they are highly porous and show a large exposed surface area that allows the light to pass through the material without substantial energy loss while offering high accessibility to electrolytes, sacrificial components, and co-catalysts; (5) they are usually stable within a large range of pH thanks to the covalent bonds between their building blocks, which is a key requisite for water splitting reactions; and (6) they are rigid, which enhances the excited state lifetime and allows for conjugation and stacking, which increase the charge mobility and prevent deactivation.
2. Crucial Aspects Determining the Photocatalytic Hydrogen Evolution Performance of COF-Mediated Systems
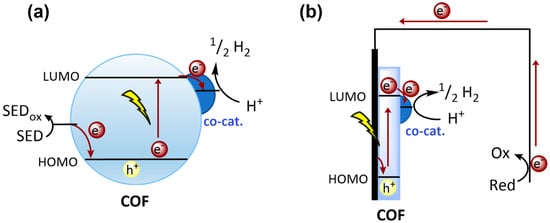
After exciton formation upon light absorption, the electron–hole pairs need to be separated into free charges and migrate to the surface of the COF to interact with the substrate and perform catalysis. In the HER case, we are interested in the capability of electrons to reduce protons into hydrogen; therefore, the CB should have a more negative potential than that of the H+/H2 couple at the working pH. Additionally, holes in the VB need to be consumed by trapping electrons (promoting an oxidation reaction) to compensate for the global charge of the semiconductor. Therefore, when the HER is studied independently from the anodic counterpart (where typically the challenging water oxidation reaction takes place), a sacrificial electron donor (SED) is needed. Thus, SED compounds such as triethanolamine (TEOA), triethylamine (TEA), diethylamine (DEA), and ascorbic acid (AA) as hole scavengers are used to consume the photogenerated holes and facilitate the global photocatalytic process. Usually, the reaction with the SED takes place before the catalytic process itself, and in consequence the former process is responsible for generating long-lived charges. In addition, this process also avoids the degradation of the photocatalyst that the accumulation of energetic holes may cause. However, before such redox processes take place at the surface of the COF, there is the possibility that the photogenerated electrons and holes recombine in the so-called charge recombination process, releasing the initial harvested energy in the form of radiation (fluorescence) or heat, thus decreasing the global performance of the whole photocatalytic process. A general overview of the photocatalytic HER mediated by COFs in a colloidal dispersion is shown in Figure 1a. Another approach that can be used to avoid the use of a SED is the deposition of the COF onto an electrode to be used as working electrode into a photoelectrochemical system. In this approach, the holes generated in the VB will be filled with electrons extracted from the oxidative reaction taking place in the counter electrode (Figure 1b).
Figure 1. General overview of the photocatalytic HER mediated by COFs in a colloidal dispersion (a) and deposited onto an electrode (b).
Despite the thermodynamic and kinetic aspects related to the photocatalytic process itself described above, the stability and fate of the whole system under turnover conditions cannot be disregarded. This includes both the organic part (COF) and the metallic (co-catalyst) part. Regarding the former, during the last 6 years it has been made clear that imine-derived
[10][18], triazine-containing
[9][11][12][13][14][9,19,20,21,22], diacetylene-containing
[15][23], β-ketoenamine-derived
[15][16][17][23,24,25], and fully π-conjugated sp
2 C
[18][19][20][26,27,28] COFs are very resistant under photocatalytic conditions, in contrast to the great tendency towards hydrolysis of boroxine and boronic ester-derived COFs
[2][21][2,29]. Additionally, N-rich COFs have arisen as highly crystalline, porous, and chemically stable materials, showing enhanced light-harvesting and charge separation properties
[22][21][23][14,29,30]. Therefore, these systems will constitute the core of this review. In contrast, the fate of co-catalysts under turnover conditions has not been paid much attention yet
[9].
2.1. Common Synthetic Pathways to Build Efficient and Robust COFs
In this section, we will give a short overview of the synthetic pathways (
Figure 2) and the building units (
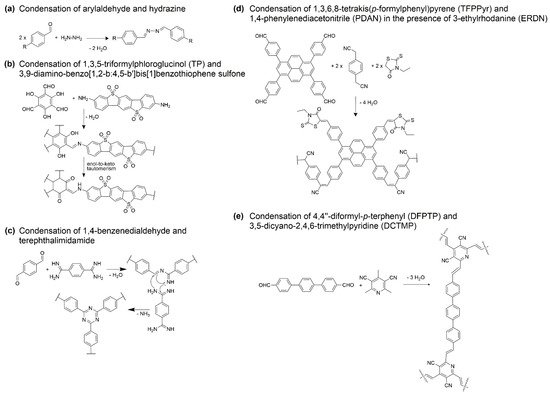
Figure 3) that give rise to COFs with remarkable HER photocatalytic properties. Condensation reactions are amongst the most used. For example, Lotsch and collaborators synthesized a series of four azine-linked triphenylaryl COFs by condensation of aldehyde groups with hydrazine (see
Figure 2a as a simple condensation example between an arylaldehyde and hydrazine to form an azine linker
[22][14]) containing a central aryl ring with different N contents, from 0 to 3 (N
x-COF, x = 0–3,
Figure 4a)
[11][19], which will be thoroughly described in
Section 2.2. Another example can be found in the condensation reaction between 2,4,6-trihydroxybenzene-1,3,5-tricarbaldehyde (also named 1,3,5-triformylphloroglucinol, TP) and 3,9-diamino-benzo[1,2-
b:4,5-
b′]bis[1]benzothiophene sulfone (
Figure 2b), involving the formation of a Schiff base between the aldehyde and the amino groups after dehydration, and the subsequent irreversible enol-to-keto conversion to form the β-ketoenamine-linked FS-COF
[16][24] (
Figure 4d) containing the sulfone functional groups (FS stands for fused sulfone). Additionally, covalent triazine framework (CTF)-based COFs are among the most relevant photocatalytic examples, whose properties will be discussed later
[24][31]. CTF-COFs can be synthesized under mild conditions by condensation at 120 °C of aldehydes (e.g., 1,4-benzenedialdehyde,
Figure 2c) and amidines (e.g., terephthalimidamide,
Figure 2c) involving the formation of a Schiff base between the aldehyde and the amidine groups after dehydration, followed by a Michael addition reaction to form the triazine ring after the elimination of ammonia (
Figure 2c)
[12][13][20,21].
Figure 2. Selected condensation reactions to achieve the synthesis of COFs. Note that for the sake of simplicity only the reactivity of one end of both reactants is shown.
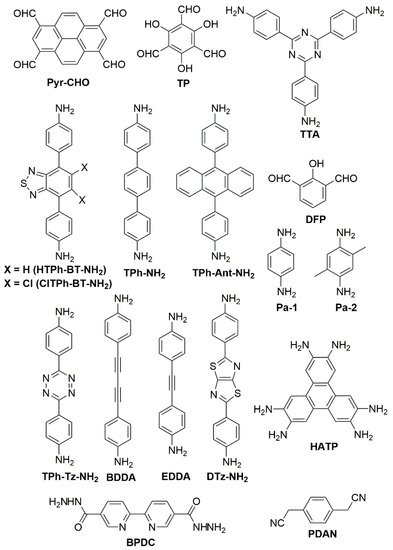
Figure 3. Structures of the starting building blocks used in some of the works.
Structures of the starting building blocks used in some of the works cited in this review.
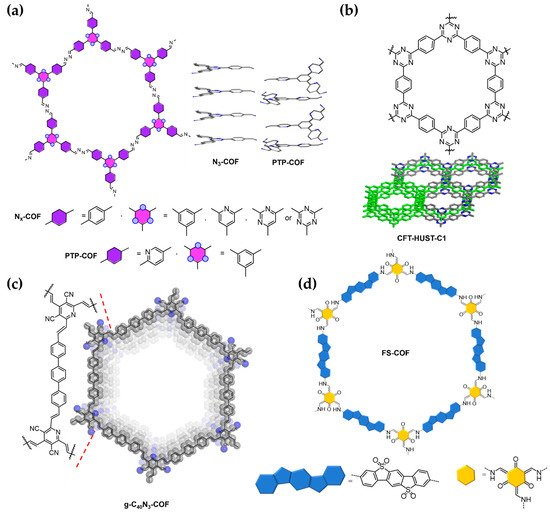
Figure 4. Highly crystalline N
x-COFs (x = 0–3) and the PTP-COF
[11][19] (
a), CTF-HUST-C1
[12][20] (
b), the g-C
40N
3-COF
[20][28] (
c), and the FS-COF
[16][24] (
d). The π-stacking interactions in the N
3-COF and PTP-COF are also displayed in (
a).
There has been a growing interest in the use of donor–acceptor (D-A) building units in COFs, for example incorporating conjugated aromatic rings (D) and cyano groups (A). In those cases, a pathway to obtain rough COFs is by C=C bond formation. Vinyl bonds are obtained, for example, by condensation between a pyrene D group (1,3,6,8-tetrakis(
p-formylphenyl)pyrene, TFPPyr) and 1,4-phenylenediacetonitrile (PDAN) in the presence of the A end-capping agent 3-ethylrhodanine (ERDN) for the formation of sp
2 c-COF
ERDN [18][26], where TFPPyr has been functionalized with ERDN at the 3,6 positions and linked at the 1,8 positions through sp
2 C-bonds to PDAN (
Figure 2d). Another remarkable example is shown in
Figure 2e, where the C=C bond condensation reaction between 4,4″-diformyl-p-terphenyl (DFPTP) and the methyl groups in 3,5-dicyano-2,4,6-trimethylpyridine (DCTMP) yielded the highly crystalline g-C
40N
3-COF
[20][28] (
Figure 4c). In this work, the authors highlight the common difficulties in obtaining highly crystalline COFs containing C=C bridges due to the poor reversibility of the traditional C=C forming reactions, and the novel synthetic strategy they develop leading to high crystallinity based on a self-healing reversible process. Other D-A building blocks that yield COFs with remarkable activity are also shown in
Figure 3. Thus, pyrene (Pyr)-containing D-A COFs have been synthesized by fusion of the D unit 4,4′,4″,4‴(pyrene-1,3,6,8-tetrayl)benzaldehyde (Pyr-CHO) with a terphenyl (TPh)-based diamine annulated with the A unit benzothiadiazole (BT)—both unmodified, HTPh-BT-NH
2, and dichlorinated, ClTPh-BT-NH
2—after condensation of the terminal aldehyde and amino groups through formation of imine bonds
[10][18].
Tuning the building blocks has also been explored for some COFs, leading to rationally improved activities. For example, Zhang et al. obtained COFs of different pore sizes and crystallinities via the condensation reaction of arylmethyl C atoms with aromatic aldehydes derived from terphenyl, biphenyl, or trisphenylbenzene
[20][28]. Due to its appropriate band energy levels, the system with a terphenyl spacer and larger pores, g-C
40N
3-COF (
Figure 2e,
Figure 4c), resulted the most active in the HER. Additionally, β-ketoenamine linked honeycomb-like COFs have been formed by condensation of 1,3,5-triformylphloroglucinol (TP) with 4,4′-diamino-
p-terphenyl (TPh-NH
2) and three derivatives consisting in replacing the central phenyl ring by anthracene (Ant), tetrazine (Tz), or benzothiadiazole (BT), namely TPh-Ant-NH
2, TPh-Tz-NH
2, and HTPh-BT-NH
2, as depicted in
Figure 3, in order to study the effect of the linker in isoreticular COFs in the overall HER photocatalytic activity
[25][32]. Another crystalline β-ketoenamine linked COF (TP-BDDA COF) has been synthesized by Thomas et al., in this case bearing diphenyl diacetylene bridges, by mixing TP and 4,4′-(buta-1,3-diyne-1,4-diyl)dianiline (BDDA) (
Figure 3)
[15][23]. The importance of the presence of the diacetylene bridges is made clear when comparing the behavior of the TP-BDDA COF (showing steady photochemical H
2 evolution for 10 h) with that of the corresponding COF with a diphenyl acetylene bridge instead (TP-EDDA COF, EDDA standing for 4,4′-(ethyne-1,2-diyl)dianiline),
Figure 3. For the latter, almost no hydrogen evolution is observed.
Other functionalization steps have been explored to favor the coordination of metals other than Pt as co-catalysts. Thus, the imine-linked HDCOF formed by reaction of 2,3,6,7,10,11-hexaaminotriphenylene (HATP) with 2,6-diformylphenol (DFP) offers N,N,O,O-tetradentate Shiff base sites to coordinate Cu(II) ions
[26][33]. Additionally, a Co-modified hydrazone-based NUS-55 COF has been synthesized by reaction of 2,2′-bipyridine-5,5′-dicarbohydrazine (BPDC) and 1,3,5-triformylphloroglucinol (TP)
[27][34]. All these building units are plotted in
Figure 3.
Finally, a recent report has demonstrated the great power of the post-functionalization strategies. In this work, a β-ketoenamine linked COF derived from TP and bipyridine (TP-Bpy-COF) was covalently modified by reaction with 1,2-dibromoethane, yielding the quaternization of some of the Bpy-bridging units. The ethylene bridge formed between two cationic adjacent N atoms (Tp-2C/Bpy
2+-COF) acts as an integrated electron mediator, resulting in a concomitant increase in photocatalytic HER activity when combined with a Pt co-catalyst
[28][35]. Another post-functionalization strategy has also been reported recently, in which a CTF-COF has been treated in HCl after having been exfoliated by ball-milling, thus generating the formation of Cl-intercalated CTF-1 through Cl-C and Cl-N covalent bonds within the triazine ring
[29][36]. The formed COF interlayer channels facilitate charge separation and mobility as well as a narrowing of the bandgap, therefore enhancing its light absorption and photocatalytic HER capabilities.
2.2. Secondary Interactions
In addition to the covalent bonds, secondary interactions, mostly π-stacking of aromatic rings, play a relevant role in the photocatalytic activity of COFs, as they govern their porosity and increase their crystallinity to a great extent. A high degree of crystallinity provides efficient pathways for charge separation and transport as well as a broader light absorption capability and higher thermal/photocatalytic stability.
A paradigmatic example is found for N
x-COFs (x = 0–3,
Figure 4a), where the crystallinity is strongly dependent on the geometrical constraints imposed by the central aromatic nodes. Thus, with increased N content the dihedral angle between the central ring and the surrounding rings decreases, increasing the planarity of the layers. This favors the crystallinity and porosity of the systems and facilitates the charge mobility through the layers and in the axial direction as well, which yields faster H
2 production and higher AQE values
[11][19]. In addition, the increase in crystallinity leads to an increase in the number of N
x-COF layers, which in turn provokes an increase in the HOMO energy level and a decrease in the LUMO one, leading to a decreased bandgap and a higher visible light absorption capability
[11][19]. Furthermore, the replacement of the central triazine ring of N
3-COF by a phenyl ring and of the terminal 4-benzaldehyde groups by 3-pyridinecarboxyaldehyde yields a phenyl-tripyridyl-derived COF (PTP-COF,
Figure 4a)
[30][37], allowing for the analysis of the differences between the two COFs containing the three N atoms at different positions. The lower symmetry of the PTP-COF compared with the N
3-COF leads to higher disorder for the former and thus lower crystallinity and porosity, since there is a higher number of possible molecular conformations around the torsion angle between the central phenyl ring and the peripheral pyridine rings than between the central triazine ring and the external phenyl substituents. In contrast to the face-to-face π-stacking interactions observed in the N
3-COF, PTP-COF crystals (
Figure 4a) are less ordered due to the interactions between the donor phenyl groups and the acceptor pyridine rings. Furthermore, the higher basicity of pyridine compared with triazine leads to H-bonding interactions between the pyridyl groups in PTP-COF crystals, leading to oligomers or molecules occluding the pores. In short, the different constitutive aromatic rings in the PTP-COF compared with the N
3-COF directly point to a lower crystallinity and porosity of the former, which shows a poorer dispersibility in water as well, with the consequent diminished light absorption capacity.
There are more examples in the literature demonstrating that the photocatalytic HER is improved with an increased crystallinity of the COF scaffold. This is the case for the two following works of Tan et al. dealing with CTF-COFs
[12][14][20,22]. In the first example
[12][20], the substitution of an aldehyde precursor by its corresponding alcohol analogue led to a decrease in the reaction rate of formation of the triazine-based COFs. The subsequent decrease in the nucleation rate results in a significant increase in the crystallinity degree of the final CTF-COFs obtained and thus of their photocatalytic HER activity with respect to CTF-COF analogues of low crystallinity. For example, when combined with Pt as co-catalyst, the crystalline CTF-HUST-C1 (
Figure 4b) displayed a 4.75-fold increase in the H
2 evolution rate than its corresponding low-crystalline analogue, CTF-HUST-1. In the second example
[14][22], the same CTF-COF was synthesized starting from the 1,4-phenylenedimethanamine precursor instead of the previous aldehyde or alcohol precursors, which in the presence of a strong base (
tBuOK) produced in a high yield (99.2%) the highly crystalline and hydrophilic (wettable) CTF-HUST-A1 COF. By using Pt as co-catalyst and TEOA as SED, this system displayed a 6.3-fold increase in the hydrogen evolution rate compared with CTF-HUST-1, that is, 9.2 mmol H
2·g
−1·h
−1, and a high AQE of 7.4% at 420 nm. Furthermore, in this work the authors also demonstrate a gradual increase in the crystallinity of the obtained CTF-HUST-A1 with the strength of the base used for its synthesis, which also correlates with a large increase in the lifetime of the photogenerated charges according to transient absorption spectroscopy. Similarly, the crystalline FS-COF (
Figure 4d) shows a 9-fold higher H
2 production than its amorphous counterpart using Pt as co-catalyst and AA as SED
[16][24], and the crystalline sp
2 c-COF shows a one order of magnitude higher photocatalytic activity than its amorphous counterpart using Pt as co-catalyst and TEOA as SED
[18][26]. Another family of highly crystalline COFs are the sp
2 C-linked ones containing
trans-disubstituted C=C bonds, among which the g-C
40N
3-COF (
Figure 2e,
Figure 4c) is the most active in the photocatalytic HER
[20][28].
COFs’ crystallinity is closely related to the stability of the systems in the photocatalytic HER. Thus, monitoring its evolution with time under turnover conditions becomes key to understanding the durability and fate of the tested systems. Thus, for example, the highly crystalline imine-linked Pyr-ClTPh-BT-COF
[10][18] and the triazine-linked N
3-COF
[11][19], possessing planar π-delocalized planes and π-π stacking interactions between parallel planes, produce H
2 under continuous irradiation at pH 7 for 2 days without any crystallinity or porosity loss. Other examples of crystalline COFs with long-term stability under photocatalytic HER conditions are the β-ketoenamine-linked FS-COF
[16][24] and the TpDTz-COF
[17][25]. These COFs (see
Section 2.5), and a recently reported thiazole (TZ)-bridged highly porous fully aromatic framework
[31][38], all exhibit photocatalytic hydrogen evolution for more than 50 h with no sign of crystallinity degradation.
There is only one exception to the rule that directly relates crystallinity and photochemical HER activity
[25][32]. In this work, Seki, Abe et al. systematically analyze the effect of all possible factors on the photocatalytic H
2 evolution activity of the β-ketoenamine linked honeycomb-like COFs TPh-Ant-NH
2, TPh-Tz-NH
2, and HTPh-BT-NH
2 (
Figure 3). The authors conclude that neither their degree of crystallinity nor the torsional angle between the external aniline rings and the central aromatic ring in TPh and derivatives are crucial for catalysis. Instead, charge mobility is facilitated by the eclipsed AA’ stacking between adjacent parallel planes compared with AB or ABC stackings, which can be obtained under different reaction conditions (solvent and temperature).
Finally, in addition to the π-stacking interactions between the aromatic rings, the secondary interactions between the COFs and the solvent, water in this case, are also key to achieving high photocatalytic activities. This is the case for the FS-COF
[16][24], where the presence of sulfone polar groups increases its effectively exposed internal surface area in front of water, thus increasing its wettability and photocatalytic activity. In consequence, under identical photocatalytic conditions, the FS-COF shows a 22-fold higher H
2 production and a superior AQE than the N
3-COF, which lacks the sulfone groups and shows reduced wettability in the reaction media.
2.3. Electronic Effects
To increase the HER photocatalytic performance of COFs, one must improve: (1) their charge separation and transfer abilities; (2) their charge generation capacity (improving light absorption by bandgap tuning); and (3) the stabilization of the key intermediates formed. All these can be achieved by tuning the electronic properties of the prepared COFs.
2.4. Heterojunctions and Encapsulation of COFs with Other Materials
A very successful alternative way to facilitate the spatial separation of photogenerated electrons and holes and thus slow down their recombination is to hybridize COFs with either metallic conductors, inorganic semiconductors, or inorganic polymers (MOFs) through the use of heterojunctions. The underlying idea consists in the good matching bandgaps of the COF and the inorganic component of the hybrid systems, favoring charge separation across the two materials. The efficient charge separation across the covalent heterojunction interface in these hybrid materials makes them greatly efficient for this photocatalytic process, yielding superior H
2 formation rates. Some significant examples are listed below: (1) the 2D/2D organic/inorganic hybrid composed of a honeycomb-like β-ketoenamine linked COF deposited in situ on top of an amine-substituted titanium carbide derivative (NH
2-Ti
3C
2T
x) through covalent Ti-O bonds
[32][43], yielding a H
2 production as high as 14.2 mmol·g
−1·h
−1 in the presence of Pt and an elevated AQE at 420 nm of 7.75%; (2) the β-ketoenamine linked COF obtained from reaction of TP with HTPh-BT-NH
2 (
Figure 3) deposited on top of g-C
3N
4, yielding 11.7 mmol H
2·g
−1·h
−1 in the absence of Pt and 26.0 H
2·g
−1·h
−1 in the presence of Pt
[33][44]; (3) CTF-HUST-C1 deposited on top of 2 nm-thick black phosphorus nanosheets through P-C covalent bonds, yielding 17.1 mmol H
2·g
−1·h
−1 in the absence of Pt
[34][45]; (4) the MOF@COF system where a 20 nm hydrazone-linked COF shell was grown on top of the octahedral NH
2-UiO-66 Zr-based MOF through imine bonds, achieving a photocatalytic H
2 evolution of 7.2 mmol·g
−1·h
−1 in the presence of Pt
[35][46]; (5) the inverse approach in which a NH
2-UiO-66 Zr-based MOF layer was deposited through imine covalent bond formation on top of a honeycomb-like β-ketoenamine linked COF (TpPa-1-COF) generated from reaction of TP with
p-phenylenediamine (Pa-1) (
Figure 3) to generate an extremely active COF@MOF hybrid system that yields 23.4 mmol H
2·g
−1·h
−1 also in the presence of Pt
[36][47]; and (6) the doping of a biphenyl-bridged CTF-based COF (CTF-HC2) with a 2D-Ni(II) pyrimidine-2-thiolate framework
[37][48].
Other strategies used have consisted in (1) the encapsulation of [Mo
3S
13]
2− anionic clusters into the channels of the positively charged honeycomb-like β-ketoenamine linked COF generated from reaction of TP (
Figure 3) with ethidium bromide, generating a remarkable HER photocatalyst able to produce 13.2 mmol H
2·g
−1·h
−1 over 18 h in the absence of Pt
[38][49]; (2) the covalent modification of graphitic carbon nitride nanosheets (GCNS) by imine covalent bonding to the imine-linked COF formed from 4,4′,4″-(1,3,5-triazine-2,4,6-triyl)trianiline (TTA) and TP (
Figure 3), yielding the extraordinary value of 46.4 mmol H
2·g
−1·h
−1 and an AQE of 31.8% at 425 nm in the presence of Pt
[39][50]; and (3) the covalent anchoring of TpPa-1-COF onto reduced graphene oxide (rGO), achieving 12.0 mmol H
2·g
−1·h
−1 in the presence of Pt
[40][51].
In addition, two very interesting systems that do not need a Pt co-catalyst to be significantly active emerge from the combination of hexagonal hematite (α-Fe
2O
3) nanosheets on top of the honeycomb-like β-ketoenamine linked TpPa-2-COF generated from reaction of TP with 2,5-dimethyl-
p-phenylenediamine (Pa-2) (
Figure 3)
[41][52], rendering 3.8 mmol H
2·g
−1·h
−1; and from the in situ growth of the TpPa-1-COF in exfoliated MoS
2, generating a hybrid system where MoS
2 is deposited on top of the TpPa-1-COF
[42][53]. Other possibilities include the deposition of CdS NPs on top COFs, such as the TpPa-2-COF
[43][54] or the CTF-1-COF
[44][55]; and the use of TiO
2 nanosheets functionalized with 3-aminopropyltriethoxysilane (APTES) to chemically bind TpPa-1-COF onto its surface by imine bond formation
[45][56], yielding 11.2 mmol H
2·g
−1·h
−1 and an AQE of 7.6% at 420 nm in the presence of Pt.
Finally, a completely different approach to promote efficient charge separation and larger photoexcitation rates as well as decreased non-radiative recombination processes consists in the insulation of the Pt NPs with a PVP-shell, which physically separates the COF from the co-catalyst but does not avoid their mutual electronic communication through electron tunneling. In the case of the TpTa-1-COF, the presence of the PVP-shell surrounding the Pt co-catalyst increases the whole photocatalytic HER efficiency up to 32 times, yielding a H
2 production of 8.4 mmol g
−1·h
−1 [46][57].
2.5. Choice of Metal Co-Catalyst
As stated before, only a few COFs have shown significant photochemical activity in the HER without the need to add a metal-based co-catalyst
[10][19][20][41][42][18,27,28,52,53]. Among the different transition metals able to promote the HER, until now very few works have used a metal co-catalyst other than Pt either in the molecular form or as NPs. Apart from enhancing the HER kinetics, co-catalysts can also reduce charge recombination by spatially separating and stabilizing charges. This highlights the need to tune the COF/co-catalyst interface to enhance the charge transfer to the co-catalyst and reduce charge recombination. A clear example is the case of non-covalently bonded molecular co-catalysts. Physically separated from the COF and working through outer-sphere electron-transfer processes, they show the competitive advantage of blocking charge recombination events. Thus, Lotsch et al. incorporated three different cobaloxime(III) co-catalysts within the azine-linked N
2-COF (
Figure 4a)
[47][58]. The same research group introduced a Ni thiolate hexameric cluster assembled in situ from a Ni(II) salt and 2-mercaptoethanol within a β-ketoenamine-linked crystalline COF bearing polar water-soluble N/S-containing 2,5-diphenyl-[1,3]-thiazolo[5,4-
d][1,3]thiazole (DTz,
Figure 3) bridges (TpDTz-COF)
[17][25]. The superior photocatalytic HER activity of the TpDTz-COF/Ni-cluster system when compared with its counterpart bearing surface-bonded Pt NPs as co-catalyst is assigned to an effective blocking of charge recombination by the physiosorbed co-catalyst
[17][25].
The presence of co-catalyst stabilizing groups on the COF’s structure has been shown to be relevant in some specific cases and is a strategy that can be further exploited to fine-tune both the morphology and intrinsic kinetics of the co-catalyst and the COF/co-catalyst interface (in relation to charge transfer and recombination processes). This is, for instance, the case for the crystalline CTF-COF containing bipyridine bridges (CTF-HC6) for which both Pd and Pt NPs were grown at its surface
[48][59]. Pd, with better affinity than Pt for the pyridylic N atoms of the COF bridges, yielded smaller NPs (2.9 nm vs. 4.3 nm in the case of Pt) at the COF surface, which resulted in superior photocatalytic HER activity (11.1 times higher for Pd than for Pt under the same experimental conditions). In a related but more recent strategy, the introduction of covalently bonded single co-catalyst atoms into the COF’s structure through synthetic covalent functionalization (generating the so-called metal-coordinated COFS, M-COFS) has also shown potential to improve charge separation and avoid recombination events. One example can be found in a binuclear Cu-containing COF, Cu-salphen-HDCOF
[26][33]. The imine-linked salphen-HDCOF has two perfect vacancies for the binding of two Cu(II) ions in a N,N,O,O-tetradentate Schiff base environment. Thus, reaction of salphen-HDCOF with Cu(II) acetate leads to the formation of Cu-salphen-HDCOF, which, after exfoliation into narrow nanosheets, acts as the co-catalyst during the photocatalytic HER. Another example can be found in the Co-modified hydrazone-based NUS-55 COF obtained by reaction of the COF with [Co(bpy)
3]Cl
2 [27][34], with a moderate photocatalytic HER activity (2.5 mmol H
2·g
−1·h
−1).
Overall, together with the general challenge of finding alternatives to precious metals as co-catalysts in the COF-based photocatalytic HER while keeping the overall performance, further research on the engineering of appropriate COF/co-catalyst interfaces is required. The combination of COF post-functionalization strategies with the tunability of both NPs through surface functionalization with stabilizing agents and molecular/single-atom catalysts through ligand design provides a myriad of combinations to rationally design better interfaces with optimized charge-transfer and separation properties.