Nuclear factor erythroid 2‐related factor 2 (NRF2) is a transcription factor that regulates the cellular defense against toxic and oxidative insults through the expression of genes involved in oxidative stress response and drug detoxification. NRF2 activation renders cells resistant to chemical carcinogens and inflammatory challenges. In addition to antioxidant responses, NRF2 is involved in other cellular processes, including metabolism and inflammation, and its functions are beyond the originally envisioned. NRF2 activity is tightly regulated through a complex transcriptional and post-translational network that enables it to orchestrate the cell’s response and adaptation to various pathological stressors for the homeostasis maintenance. Elevated or decreased NRF2 activity by pharmacological and genetic manipulations of NRF2 activation is associated with many metabolism- or inflammation-related diseases. Emerging evidence shows that NRF2 lies at the center of a complex regulatory network and establishes NRF2 as a truly pleiotropic transcription factor. Here we summarize the complex regulatory network of NRF2 activity and its roles in metabolic reprogramming, unfolded protein response, proteostatsis, autophagy, mitochondrial biogenesis, inflammation, and immunity.
- NRF2, INFLAMMATION, IMMUNITY, CANCER METABOLISM
1. Introduction
Homeostasis is key to organismal health and survival. Environmental stress is ubiquitous and unavoidable to all living beings and threatens to disrupt cell functions. Organisms respond and adapt to stresses through defined regulatory mechanisms. The transcription factor nuclear factor erythroid 2-related factor 2 (NRF2) is best known as one of the main orchestrators of the cellular xenobiotic and oxidative stress response. NRF2, encoded by the gene nuclear factor, erythroid 2 like 2 (NFE2L2), belongs to the Cap’n’Collar (CNC) subfamily of basic leucine zipper (bZIP) transcription factors, which comprises nuclear factor erythroid-derived 2 (NFE2) and NRF1, NRF2, and NRF3 [1]. NRF2 possesses seven conserved NRF2-ECH homology (Neh) domains with different functions to control NRF2 transcriptional activity (Figure 1A). The bZip in the Neh1 domain heterodimerizes with small musculoaponeurotic fibrosarcoma proteins (sMAF) K, G, and F as well as other bZip proteins to recognize antioxidant response elements (ARE) for activation of gene transcription, whereas the Neh2 domain contains ETGE and DLG motifs that specifically interact with Kelch domain of Kelch-like-ECH-associated protein 1 (KEAP1) to mediate NRF2 ubiquitination and degradation (Figure 1B) [2]. The Neh3-5 domains function as transcriptional activation domains by binding to various components of the transcriptional machinery [3]. Neh6 domain contains two redox-independent degrons DSGIS and DSAPGS that bind to E3 ubiquitin ligase β-transducin repeat-containing protein (βTrCP), which mediates NRF2 degradation in oxidatively stressed cells (Figure 1C) [4]. Neh7 domain mediates interaction with retinoic X receptor alpha (RXRα), which represses NRF2 activity [5]. These domains modulate NRF2 stability and transcriptional activation of its target genes at multiple levels, including transcriptional and post-transcriptional and post-translational regulation in response to various insults. Recent studies have identified new NRF2 target genes and revealed several new functions of NRF2 that go beyond its redox-regulating capacities, including regulation of inflammation, autophagy, metabolism, proteostasis, and unfolded protein response (UPR), particularly in the context of carcinogenesis. NRF2 has become a prime subject of extensive research involving inflammation, metabolism, cancer prevention and treatment, and its functions are more far-reaching than originally envisioned. Understanding the regulation of NRF2 activity and its new emerging functions presents new challenges but also new opportunities for targeting NRF2 in cancer.
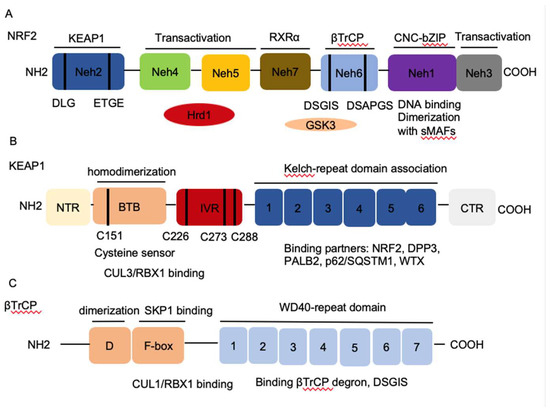
Figure 1. The architecture of Nuclear factor erythroid 2-related factor 2 (NRF2), Kelch-like-ECH-associated protein 1 (KEAP1), and β-transducin repeat-containing protein (βTrCP). (A) NRF2 contains seven conserved NRF2-ECH homology NRF2-ECH homology (Neh) domains, Neh1-Neh7. Neh1 contains a basic leucine zipper (bZip) motif, where the basic region is responsible for DNA binding and the Zip dimerizes with other binding partners such as sMAFs. Neh2 contains ETGE and DLG motifs, which are required for the interaction with KEAP1 and subsequent KEAP1-mediated proteasomal degradation. Neh3, 4 and 5 domains are transactivation domains of NRF2. Neh4 and 5 domains also interact with HRD1 that mediates NRF2 degradation. Neh6 contains two βTrCP degrons DSGIS and DSAPGS that are responsible for the β-TrCP mediated proteasomal degradation. (B) KEAP1 contains five domains, amino terminal region (NTR), a broad complex, tramtrack, bric-a-brac (BTB) domain, an intervening region (IVR), six Kelch domains, and the C-terminal region (CTR). The Kelch domain and CTR mediate the interaction with NRF2, p62, DPP3, WTX, and PALB2 that contains ETGE motifs. The BTB domain homodimerizes with KEAP1 and contributes to the interaction of IVR with Cul3/RBX1 complex. Several functional important cysteine residues (C151, C226, C273 and C278) that sense reactive oxygen species (ROS) and electrophiles and modulate KEAP1-NRF2 interaction. (C) βTrCP has three domains, dimerization domain (D) that forms homo- and heterodimers between βTrCP1 and βTrCP2, the F-box that recruits SKP1 for the binding of CUL1/RBX1 complex, and the WD40 repeat domain that binds βTrCP degrons DSGIS and DSAPGS in NRF2. βTrCP, β-transducing repeat-containing protein; CUL3, Cullin3; RBX1, RING-box protein; WD40, WD Repeat protein 40; RXRα, retinoic X receptor alpha; DPP3, dipeptidyl peptidase 3; WTX, Wilms tumor gene on X chromosome; PALB2, Partner and Localizer of BRCA2; GSK3, Glycogen synthase kinase-3; SKP1, S-phase kinase-associated protein-1.
2. Regulation of NRF2 Activity
2.1. Regulation of NRF2 Protein Stability
NRF2 abundance within the cell is tightly regulated and is mainly controlled by four E3 ubiquitin ligase complexes-mediated ubiquitylation and proteasomal degradation: KEAP1-Cullin (CUL) 3-RING-box protein (RBX)1, βTrCP-S-phase kinase-associated protein-1 (SKP1)-CUL1-RBX1, WD Repeat protein (WDR), 3-CUL4-damaged DNA binding protein (DDB) 1, and HRD1 (also called Synoviolin) under different conditions. NRF2 is expressed in all cell types and its basal protein levels are usually low in unstressed conditions mainly due to KEAP1 mediated proteasomal degradation (Figure 2). KEAP1 is a redox-regulated adaptor for the CUL3- RBX1 ubiquitin ligase complex and it binds NRF2 through its C-terminal Kelch domain, which interacts with the DLG and ETGE motifs in the Neh2 domain of NRF2, resulting in ubiquitination of NRF2 in the cytoplasm and degradation by the 26 S proteasome [3]. This constitutive degradation of NRF2 ensures that only the basal expression of NRF2 target genes are maintained for housekeeping functions. During oxidative stress, electrophiles and reactive oxygen species (ROS) react with sensor cysteines of KEAP1, including cysteine 151 (C151), C273, and C288, to allow NRF2 to escape from KEAP1 mediated degradation [6][7]. As a result, newly synthesized NRF2 accumulates in the nucleus and activates the expression of cytoprotective genes [3][8]. In addition, KEAP1 dependent but cysteine independent mechanisms were reported to stabilize NRF2 by interfering with formation of the NRF2-KEAP1 complex. Autophagy cargo-adaptor p62/sequestosome 1 (SQSTM1) [9][10][11], dipeptidyl peptidase 3 (DPP3) [12], Wilms tumor gene on X chromosome (WTX) [13], and Partner and Localizer of BRCA2 (PALB2) [14] all contain KEAP1-interacting region (KIR)-like ETGE motifs and thus competes with NRF2 for KEAP1 binding, resulting in KEAP1 sequestration and NRF2 stabilization. p21Cip1/WAF1 [15] and Breast Cancer Type 1 Susceptibility Protein (BRCA1) [16] can also directly interact with the ETGE motif of NRF2, thus competes with KEAP1 for NRF2 binding and stabilizes NRF2. Furthermore, the acetyltransferase p300 stabilizes NRF2 and increases its nuclear localization by interfering KEAP1-NRF2 interaction [17].
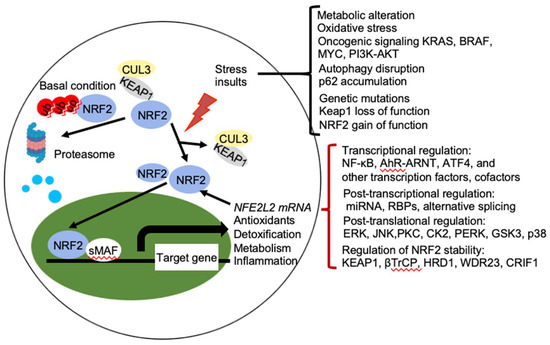
Figure 2. Regulation of NRF2 activity. Under basal conditions, the amount of NRF2 is low due to its continuous sequestration by KEAP1 and subsequent proteasomal degradation. Under stressed condition, the cellular NRF2 amount is temporarily or constitutively increased upon exposure to toxicants and ROS, oncogenic signaling, genetic mutations, autophagy disruption, or metabolic alteration, which disrupt the KEAP1-NRF2 complex and lead to activation of NRF2. Activated NRF2 accumulates in the nucleus, where it interacts with other transcription factors and cofactors to regulate transcription of its target genes, which encoding proteins involved in the antioxidants, detoxification, metabolism, and inflammation. NRF2 also regulates its own NFE2L2 mRNA transcription. Activity of NRF2 is modulated at multiple levels, including transcriptional regulation (NF-κB, AhR-ARNT, ATF4, and other transcription factors, cofactors), post-transcriptional regulation (miRNA, RBPs, alternative splicing), post-translational regulation (ERK, JNK, PKC, CK2, PERK, GSK3, p38), and regulation of NRF2 stability (KEAP1, βTrCP, HRD1, WDR23, CRIF1). AhR, aryl hydrocarbon receptor; ARNT, AHR nuclear translocator; NF-κB, nuclear factor-κB; PI3K, phopshoinositide 3-kinase; PKC, protein kinase C; ERG, extracellular signal-regulated protein kinases; JNK, c-jun N-terminal kinase; PERK, protein kinase R (PKR)-like endoplasmic reticulum kinase; CK2, casein kinase 2; WDR23, WD40-repeat protein 23; CRIF1, CR6-interacting Factor 1; ATF4, activating transcription factor.
In contrast to KEAP1-CUL3-RBX1 mediated degradation under basal conditions, βTrCP-SKP1-CUL1-RBX1 and WDR3-CUL4-DDB1 can contribute to NRF2 degradation under both basal and oxidative stress conditions, independent of KEAP1 [18][19][20]. NRF2 contains two motifs, DSGIS and DSAPGS in its Neh6 domain, which can be recognized by the F box of the WD40 substrate adaptor βTrCP. Glycogen synthase kinase-3 (GSK3) phosphorylates the DSGIS motif and increases the affinity of βTrCP for NRF2, thereby stimulating NRF2 ubiquitination and degradation [2][18][19]. GSK3 is inhibited by phosphorylation of an N-terminal serine residue (Ser9 in GSK-3β and Ser21 GSK-3α, respectively) by protein kinase B (PKB)/AKT. The signaling that activates upstream phosphoinositide 3-kinase (PI3K)-PKB/AKT, including growth factors, mTOR, and oncogenic RAS signaling, inhibits GSK3 and thereby stabilizes NRF2 by suppressing βTrCP-mediated degradation [19]. WDR23 is a WD40-repeat protein and it binds the DIDLID sequence within the Neh2 domain of NRF2 to regulate NRF2 ubiquitination and degradation [20]. HRD1 is an endoplasmic reticulum (ER) membrane-associated E3 ubiquitin ligase involved in ER-associated degradation (ERAD). Under ER stress conditions, HRD1 is induced and binds to the Neh 4-5 domains of NRF2 that mediate NRF2 ubiquitylation and degradation in cirrhotic livers [21]. In addition, CR6-interacting Factor 1 (CRIF1), also known as growth arrest and DNA damage-inducible proteins-interacting protein 1 (GADD45GIP1), interacts with Neh2 domain and the C-terminal region containing Neh1 and Neh3 of NRF2 to promote NRF2 ubiquitylation and degradation in a redox-independent manner [22]. Interestingly, KEAP1-mediated NRF2 ubiquitination and degradation mainly act in the cytoplasm, whereas KEAP-1 independent NRF2 stability regulation is both cytoplasmic and nuclear, which contributes to termination of NRF2-mediated transcriptional response [20][22][23]. Furthermore, posttranslational modifications of NRF2 contribute to the changes in its binding partners and cellular localization that regulate NRF2 stability. Extracellular signal-regulated protein kinases (ERK) [24], c-jun N-terminal kinase (JNK) [24], PI3K-AKT [19], protein kinase C (PKC) [25], casein kinase 2 (CK2) [26], and protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK) [27] were reported to mediate phosphorylation of NRF2 and increase its stability and subsequent transcriptional activity, whereas p38 and GSK3-mediated phosphorylation of NRF2 decreases NRF2 stability [19]. In hepatocellular carcinoma triggered by MYC and KEAP1 inactivation, fructosamine-3-kinase (FN3K), a kinase that triggers protein de-glycation, mediates NRF2 de-glycation that stabilizes NRF2 and executes its oncogenic function [28].
2.2. Regulation of NRF2 Transcription
Transcription factor NRF2 binds to ARE in the promoter region of many cell defense genes to activate their transcription. NFE2L2 gene contains ARE within its promoter region that renders NRF2 the ability to directly activate its own transcription, providing a positive feedback mechanism to amplify NRF2 effects [29]. In addition, NFE2L2 transcription is regulated by several transcription factors, including aryl hydrocarbon receptor (AhR) [30] and nuclear factor (NF)-κB [31]. The NFE2L2 gene contains one xenobiotic-responsive element (XRE)-like element at position -712 of the promoter region and two additional XRE-like elements located at +755 and +850 that are activated by the transcription factor aryl hydrocarbon receptor (AHR) [30]. When activated by ligands, such as TCDD (2,3,7,8-tetrachlorodibenzo-p-dioxin), AHR dimerizes with AHR nuclear translocator (ARNT) to bind to the XRE of NFE2L2 and activates NRF2 transcription [30]. In addition, NFE2L2 promoter contains an NF-κB binding site, which allows it to be regulated by NF-κB [32]. Lipopolysaccharide (LPS) treatment in human monocytes induced NFE2L2 transcription [33]. Constitutive NF-κB activation mediates upregulation of NFE2L2 gene and contributes to high basal NRF2 activity that renders resistance to chemotherapy in acute myeloid leukemia (AML) [32]. Oncogenic signaling pathways including oncogenic oncogenes K-RAS(G12D), B-RAF (V619E) and MYC (ERT2) [34], the PI3K-AKT pathway [35], and the Notch signaling pathway [36], have been reported to augment NFE2L2 transcription to stably elevate the basal NRF2 antioxidant program and support its pro-tumorigenic effects. Some hypermethylation or single nucleotide polymorphisms (SNPs) of the NFE2L2 promoter region decreases NRF2 mRNA expression [26][37][38].
2.3. Post-Transcriptional Regulation of NRF2
microRNAs (miRNAs) are endogenous single-stranded, noncoding RNAs with an average 22 nucleotides in length that repress gene expression by sequence-specific binding with mRNA molecules and subsequent inhibition of protein translation and destabilization of mRNA [39]. miRNAs that have been shown to be involved in the regulation of NRF2 include miR-144 [40], miR-28 [41], miR-34 [42], miR-93 [43], and miR-153 [44]. Increased miR-144 is associated with reduced NRF2 transcriptional activity and impaired oxidative stress tolerance in erythroid cells, which is associated with the sickle cell disease [40]. miR-34 expression is increased in aging rat liver, which targets NRF2 and downstream oxidative stress defense protein Mgst1s to inhibit their expression [42]. Expression of miR-28 [41], miR-93 [43], and miR-153 [44] in breast cancer cells were reported to facilitate the degradation of NRF2 mRNA. In neuroblastoma cells, miR-153, miR-27a, miR142-5p, and miR-144 directly target 3′-untranslated region (UTR) of NFE2L2 and changes in these miRNAs either individually or as a group could result in inefficient transactivating ability of NRF2 [45]. However, validation in physiological and pathological conditions is still lacking.
In contrast to miRNAs, RNA-binding proteins (RBPs) are typically considered as proteins that bind RNA through one or multiple globular RNA-binding domains (RBDs) and change the fate or function of the bound RNAs [46]. Two of RBPs, HuR and AUF1 binds to the 3′-UTR of NFE2L2 mRNA and result in the elevated NRF2 activation [47]. HuR enhances NFE2L2 mRNA maturation and promotes its nuclear export, whereas AUF1 stabilizes NFE2L2 mRNA [47]. Alternative splicing is another mechanism that can regulate NRF2 activity. Aberrant NFE2L2 transcript variants lacking exon 2, or exons 2 and 3, have been observed in lung and head and neck cancers and the NRF2 protein isoforms encoded by these splice variants lack the KEAP1 interaction domain, thus resulting in NRF2 stabilization and induction of the NRF2 program [48]. The impact of post-transcriptional regulation of NFE2L2 on the pathogenesis of disease remains to be evaluated.
2.4. Regulation of NRF2 Transcriptional Activation of Its Target Genes
NRF2 activity is tightly regulated. Gene transcription profiles revealed that not all genes in the vicinity of bound NRF2 are transcriptionally regulated by NRF2 binding [49][50]. These genes require the recruitment of both NRF2 and NRF2 binding partners including transcription factors, cofactors, and mediators for a complete activation. sMAF transcription factors, including MAFF, MAFG, and MAFK, binds to NRF2 through their leucine zipper domains to form NRF2-MAF complexes for the recognition of ARE and supports NRF2-mediated transcriptional activation of antioxidant and metabolic genes [51]. The transcription factor BTB and CNC homology 1 (BACH1), which is essential in controlling heme level through the inhibition of heme oxygenase-1 (HO)-1 gene, competes with NRF2-sMAF interaction at ARE sites for transcriptional repression of NRF2 target NAD(P)H:quinone oxidoreductase1 (NQO1) [52]. NRF2-induced HO-1 expression requires inactivation of BACH1 [53]. Activating transcription factor 4 (ATF4) can dimerize with NRF2 at ARE to induce expression of HO-1 [54]. Activator protein 1 (AP-1) subunit c-Jun can dimerize with NRF2 and activate NRF2-induced transcription, while another AP-1 subunit c-Fos can suppress NRF2-induced transcription [55][56].
CREB-binding protein (CBP) and its cofactor p300 can be co-recruited to ARE by NRF2 through its Neh4/5 domains of NRF2 for transcriptional activation [57]. CBP and p300 are histone acetyltransferases to facilitate chromatin decondensation and the recruitment of the transcription machinery [58]. However, ATF3 can compete with CBP for the binding of NRF2 and inhibit the transcription induced by NRF2–CBP complex [59]. Interestingly, ATF3 deletion increases NRF2 degradation through upregulating KEAP1 expression and loss of DJ-1 pathways in human bronchoalveolar epithelial cells [60]. Therefore, ATF3 can either positively or negatively control NRF2 activity. Transcriptional repressor silencing mediator for retinoid and thyroid hormone receptor (SMRT) that mediates histone deacetylation, can interact with NRF2 Neh4/5 domains and inhibit NRF2-induced GSTA2 expression [61]. In addition to histone-modifying enzymes, NRF2 interacts with other co-activators of the transcription machinery, such as ATPase subunit of the SWI/SNF chromatin-remodeling complex Brahma-related gene 1 (BRG-1) [62], chromodomain helicase DNA-binding protein 6 (CHD6) [63], receptor-associated co-activator 3 (RAC3) [64], and NAD+-dependent histone deacetylase sirtuin 6 (SIRT6) [65], mediator of RNA polymerase II transcription subunit 16 [66] to selectively influence the transcription of NRF2 target genes. However, the functional significance of these interactions has not been extensively elucidated. Nuclear receptors PPARγ [67] and estrogen receptor α (ERα) [68] can directly bind to NRF2 and suppress the NRF2 activity. Interestingly, a gene dose response study analyzing expression changes in livers from Nfe2l2-null, wild-type, Keap1-knockdown, and Keap1-knockout mice showed that the extent of NRF2 transactivation depends on the levels of NRF2 protein (184). Complete understanding of NRF2-mediated gene transactivation requires paying attention to the cooperation or competition with other transcription factors and co-factors at the ARE and ARE-like sites.
3. NRF2 Regulates Autophagy
Autophagy is a protein and organelle quality control mechanism that orderly degrades and recycles cellular components, including protein aggregates and old or damaged organelles. Beside ubiquitin proteasome system, oxidative, proteotoxic, and metabolic stresses increase autophagy and help to restore homeostasis [69]. Autophagy requires the coordinated participation of a set of proteins that participate in the formation of autophagosomes and autolysosomes, as well as cargo-selective proteins that recognize specific cargos and direct them to degradation. NRF2 induces the expression of autophagy genes encoding SQSTM1/p62, calcium-binding and coiled-coil domain-containing protein 2 (CALCOCO2/NDP52), unc-51-like kinase 1 (ULK1), autophagy protein 5 (ATG5), and gamma-aminobutyric acid receptor-associated protein-like 1 (GABARAPL1) and enhances autophagy [70]. In the NRF2 activated context, autophagy-targeted therapy could be ineffective. Interestingly, insufficient autophagy leads to accumulation of oxidized proteins or organelles that can lead to NRF2 activation. More importantly, autophagy deficiency leads to accumulation of p62, a multifunctional cargo receptor that can sequester KEAP1 and stabilize NRF2, resulting in NRF2 activation [10]. Thus, p62 and NRF2 create a positive feedback loop to regulate a plethora of cellular functions [9][71].
4. Concluding Remarks and Perspectives
Although NRF2 is best known as an oxidant stress response transcription factor, it is also critical to the regulation of metabolism, inflammation, autophagy, proteostasis, mitochondrial physiology, and immune responses. NRF2-mediated target gene expression is determined by the activating stimulus, the cellular context, the availability of binding partners, interaction with other transcription factors, activators, and repressors, as well as the crosstalk with other signaling pathways. NRF2 lies at the center of a complex regulatory network and most of functions exerted by NRF2 are interconnected that contribute to the initiation and development of many diseases, particularly metabolism- or inflammation-associated diseases. Generally, NRF2 activation plays a protective role in physiological conditions, but it promotes cancer development after cancer is established. In addition, NRF2 is generally anti-inflammation, however, it exhibits opposite roles during different virus infections. Future studies need to address how the NRF2-dependent network changes from a physiological (beneficial) to a pathological (adversary) condition that can provide insights into mechanisms of disease pathogenesis. The key switch between the tumor suppressive role and tumor promoting roles of NRF2 could be due to the active NRF2 protein levels/doses and the duration of NRF2 activation in the crosstalk with specific factors and signaling pathways. In addition, due to the broad action spectrum, understanding more dynamic and integrated NRF2-mediated metabolism and immunity of different cell types in a disease context will facilitate discovery of molecular pathology and new treatment strategies.
References
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191.
- Taguchi, K.; Yamamoto, M. The KEAP1-NRF2 System in Cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef]
- Canning, P.; Sorrell, F.J.; Bullock, A.N. Structural basis of Keap1 interactions with Nrf2. Free Radic. Biol. Med. 2015, 88, 101–107.
- McMahon, M.; Thomas, N.; Itoh, K.; Yamamoto, M.; Hayes, J.D. Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J. Biol. Chem. 2004, 279, 31556–31567.
- Wang, H.; Liu, K.; Geng, M.; Gao, P.; Wu, X.; Hai, Y.; Li, Y.; Li, Y.; Luo, L.; Hayes, J.D.; et al. RXRalpha inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res. 2013, 73, 3097–3108.
- McMahon, M.; Lamont, D.J.; Beattie, K.A.; Hayes, J.D. Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proc. Natl. Acad. Sci. USA 2010, 107, 18838–18843.
- Baird, L.; Lleres, D.; Swift, S.; Dinkova-Kostova, A.T. Regulatory flexibility in the Nrf2-mediated stress response is conferred by conformational cycling of the Keap1-Nrf2 protein complex. Proc. Natl. Acad. Sci. USA 2013, 110, 15259–15264.
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; O’Connor, T.; Yamamoto, M. Keap1 regulates both cytoplasmic-nuclear shuttling and degradation of Nrf2 in response to electrophiles. Genes Cells 2003, 8, 379–391.
- Taniguchi, K.; Yamachika, S.; He, F.; Karin, M. p62/SQSTM1-Dr. Jekyll and Mr. Hyde that prevents oxidative stress but promotes liver cancer. FEBS Lett. 2016, 590, 2375–2397.
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223.
- Umemura, A.; He, F.; Taniguchi, K.; Nakagawa, H.; Yamachika, S.; Font-Burgada, J.; Zhong, Z.; Subramaniam, S.; Raghunandan, S.; Duran, A.; et al. p62, Upregulated during Preneoplasia, Induces Hepatocellular Carcinogenesis by Maintaining Survival of Stressed HCC-Initiating Cells. Cancer Cell 2016, 29, 935–948.
- Hast, B.E.; Goldfarb, D.; Mulvaney, K.M.; Hast, M.A.; Siesser, P.F.; Yan, F.; Hayes, D.N.; Major, M.B. Proteomic analysis of ubiquitin ligase KEAP1 reveals associated proteins that inhibit NRF2 ubiquitination. Cancer Res. 2013, 73, 2199–2210.
- Camp, N.D.; James, R.G.; Dawson, D.W.; Yan, F.; Davison, J.M.; Houck, S.A.; Tang, X.; Zheng, N.; Major, M.B.; Moon, R.T. Wilms tumor gene on X chromosome (WTX) inhibits degradation of NRF2 protein through competitive binding to KEAP1 protein. J. Biol. Chem. 2012, 287, 6539–6550.
- Ma, J.; Cai, H.; Wu, T.; Sobhian, B.; Huo, Y.; Alcivar, A.; Mehta, M.; Cheung, K.L.; Ganesan, S.; Kong, A.N.; et al. PALB2 interacts with KEAP1 to promote NRF2 nuclear accumulation and function. Mol. Cell. Biol. 2012, 32, 1506–1517.
- Chen, W.; Sun, Z.; Wang, X.J.; Jiang, T.; Huang, Z.; Fang, D.; Zhang, D.D. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol. Cell 2009, 34, 663–673.
- Gorrini, C.; Baniasadi, P.S.; Harris, I.S.; Silvester, J.; Inoue, S.; Snow, B.; Joshi, P.A.; Wakeham, A.; Molyneux, S.D.; Martin, B.; et al. BRCA1 interacts with Nrf2 to regulate antioxidant signaling and cell survival. J. Exp. Med. 2013, 210, 1529–1544.
- Ganner, A.; Pfeiffer, Z.C.; Wingendorf, L.; Kreis, S.; Klein, M.; Walz, G.; Neumann-Haefelin, E. The acetyltransferase p300 regulates NRF2 stability and localization. Biochem. Biophys. Res. Commun. 2020, 524, 895–902.
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781.
- Hayes, J.D.; Chowdhry, S.; Dinkova-Kostova, A.T.; Sutherland, C. Dual regulation of transcription factor Nrf2 by Keap1 and by the combined actions of beta-TrCP and GSK-3. Biochem. Soc. Trans. 2015, 43, 611–620.
- Lo, J.Y.; Spatola, B.N.; Curran, S.P. WDR23 regulates NRF2 independently of KEAP1. PLoS Genet. 2017, 13, e1006762.
- Wu, T.; Zhao, F.; Gao, B.; Tan, C.; Yagishita, N.; Nakajima, T.; Wong, P.K.; Chapman, E.; Fang, D.; Zhang, D.D. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev. 2014, 28, 708–722.
- Kang, H.J.; Hong, Y.B.; Kim, H.J.; Bae, I. CR6-interacting factor 1 (CRIF1) regulates NF-E2-related factor 2 (NRF2) protein stability by proteasome-mediated degradation. J. Biol. Chem. 2010, 285, 21258–21268.
- He, F.; Antonucci, L.; Karin, M. NRF2 as a regulator of cell metabolism and inflammation in cancer. Carcinogenesis 2020.
- Yuan, X.; Xu, C.; Pan, Z.; Keum, Y.S.; Kim, J.H.; Shen, G.; Yu, S.; Oo, K.T.; Ma, J.; Kong, A.N. Butylated hydroxyanisole regulates ARE-mediated gene expression via Nrf2 coupled with ERK and JNK signaling pathway in HepG2 cells. Mol. Carcinog. 2006, 45, 841–850.
- Huang, H.C.; Nguyen, T.; Pickett, C.B. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem. 2002, 277, 42769–42774.
- Apopa, P.L.; He, X.; Ma, Q. Phosphorylation of Nrf2 in the transcription activation domain by casein kinase 2 (CK2) is critical for the nuclear translocation and transcription activation function of Nrf2 in IMR-32 neuroblastoma cells. J. Biochem. Mol. Toxicol. 2008, 22, 63–76.
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 2003, 23, 7198–7209.
- Sanghvi, V.R.; Leibold, J.; Mina, M.; Mohan, P.; Berishaj, M.; Li, Z.; Miele, M.M.; Lailler, N.; Zhao, C.; De Stanchina, E.; et al. The Oncogenic Action of NRF2 Depends on De-glycation by Fructosamine-3-Kinase. Cell 2019, 178, 807–819.e21.
- Kwak, M.K.; Itoh, K.; Yamamoto, M.; Kensler, T.W. Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: Role of antioxidant response element-like sequences in the nrf2 promoter. Mol. Cell. Biol. 2002, 22, 2883–2892.
- Miao, W.; Hu, L.; Scrivens, P.J.; Batist, G. Transcriptional regulation of NF-E2 p45-related factor (NRF2) expression by the aryl hydrocarbon receptor-xenobiotic response element signaling pathway: Direct cross-talk between phase I and II drug-metabolizing enzymes. J. Biol. Chem. 2005, 280, 20340–20348.
- Nair, S.; Doh, S.T.; Chan, J.Y.; Kong, A.N.; Cai, L. Regulatory potential for concerted modulation of Nrf2- and Nfkb1-mediated gene expression in inflammation and carcinogenesis. Br. J. Cancer 2008, 99, 2070–2082.
- Rushworth, S.A.; Zaitseva, L.; Murray, M.Y.; Shah, N.M.; Bowles, K.M.; MacEwan, D.J. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-kappaB and underlies its chemo-resistance. Blood 2012, 120, 5188–5198.
- Rushworth, S.A.; MacEwan, D.J.; O’Connell, M.A. Lipopolysaccharide-induced expression of NAD(P)H:quinone oxidoreductase 1 and heme oxygenase-1 protects against excessive inflammatory responses in human monocytes. J. Immunol. 2008, 181, 6730–6737.
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109.
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79.
- Wakabayashi, N.; Skoko, J.J.; Chartoumpekis, D.V.; Kimura, S.; Slocum, S.L.; Noda, K.; Palliyaguru, D.L.; Fujimuro, M.; Boley, P.A.; Tanaka, Y.; et al. Notch-Nrf2 axis: Regulation of Nrf2 gene expression and cytoprotection by notch signaling. Mol. Cell. Biol. 2014, 34, 653–663.
- Yu, S.; Khor, T.O.; Cheung, K.L.; Li, W.; Wu, T.Y.; Huang, Y.; Foster, B.A.; Kan, Y.W.; Kong, A.N. Nrf2 expression is regulated by epigenetic mechanisms in prostate cancer of TRAMP mice. PLoS ONE 2010, 5, e8579.
- Suzuki, T.; Shibata, T.; Takaya, K.; Shiraishi, K.; Kohno, T.; Kunitoh, H.; Tsuta, K.; Furuta, K.; Goto, K.; Hosoda, F.; et al. Regulatory nexus of synthesis and degradation deciphers cellular Nrf2 expression levels. Mol. Cell. Biol. 2013, 33, 2402–2412.
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. (Lausanne) 2018, 9, 402.
- Sangokoya, C.; Telen, M.J.; Chi, J.T. microRNA miR-144 modulates oxidative stress tolerance and associates with anemia severity in sickle cell disease. Blood 2010, 116, 4338–4348.
- Yang, M.; Yao, Y.; Eades, G.; Zhang, Y.; Zhou, Q. MiR-28 regulates Nrf2 expression through a Keap1-independent mechanism. Breast Cancer Res. Treat. 2011, 129, 983–991.
- Li, N.; Muthusamy, S.; Liang, R.; Sarojini, H.; Wang, E. Increased expression of miR-34a and miR-93 in rat liver during aging, and their impact on the expression of Mgst1 and Sirt1. Mech. Ageing Dev. 2011, 132, 75–85.
- Singh, B.; Ronghe, A.M.; Chatterjee, A.; Bhat, N.K.; Bhat, H.K. MicroRNA-93 regulates NRF2 expression and is associated with breast carcinogenesis. Carcinogenesis 2013, 34, 1165–1172.
- Wang, B.; Teng, Y.; Liu, Q. MicroRNA-153 Regulates NRF2 Expression and is Associated with Breast Carcinogenesis. Clin. Lab. 2016, 62, 39–47.
- Narasimhan, M.; Patel, D.; Vedpathak, D.; Rathinam, M.; Henderson, G.; Mahimainathan, L. Identification of novel microRNAs in post-transcriptional control of Nrf2 expression and redox homeostasis in neuronal, SH-SY5Y cells. PLoS ONE 2012, 7, e51111.
- Hentze, M.W.; Castello, A.; Schwarzl, T.; Preiss, T. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018, 19, 327–341.
- Poganik, J.R.; Long, M.J.C.; Disare, M.T.; Liu, X.; Chang, S.H.; Hla, T.; Aye, Y. Post-transcriptional regulation of Nrf2-mRNA by the mRNA-binding proteins HuR and AUF1. FASEB J. 2019, 33, 14636–14652.
- Goldstein, L.D.; Lee, J.; Gnad, F.; Klijn, C.; Schaub, A.; Reeder, J.; Daemen, A.; Bakalarski, C.E.; Holcomb, T.; Shames, D.S.; et al. Recurrent Loss of NFE2L2 Exon 2 Is a Mechanism for Nrf2 Pathway Activation in Human Cancers. Cell Rep. 2016, 16, 2605–2617.
- Malhotra, D.; Portales-Casamar, E.; Singh, A.; Srivastava, S.; Arenillas, D.; Happel, C.; Shyr, C.; Wakabayashi, N.; Kensler, T.W.; Wasserman, W.W.; et al. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res. 2010, 38, 5718–5734.
- Chorley, B.N.; Campbell, M.R.; Wang, X.; Karaca, M.; Sambandan, D.; Bangura, F.; Xue, P.; Pi, J.; Kleeberger, S.R.; Bell, D.A. Identification of novel NRF2-regulated genes by ChIP-Seq: Influence on retinoid X receptor alpha. Nucleic Acids Res. 2012, 40, 7416–7429.
- Hirotsu, Y.; Katsuoka, F.; Funayama, R.; Nagashima, T.; Nishida, Y.; Nakayama, K.; Engel, J.D.; Yamamoto, M. Nrf2-MafG heterodimers contribute globally to antioxidant and metabolic networks. Nucleic Acids Res. 2012, 40, 10228–10239.
- Dhakshinamoorthy, S.; Jain, A.K.; Bloom, D.A.; Jaiswal, A.K. Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H:quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J. Biol. Chem. 2005, 280, 16891–16900.
- Reichard, J.F.; Motz, G.T.; Puga, A. Heme oxygenase-1 induction by NRF2 requires inactivation of the transcriptional repressor BACH1. Nucleic Acids Res. 2007, 35, 7074–7086.
- He, C.H.; Gong, P.; Hu, B.; Stewart, D.; Choi, M.E.; Choi, A.M.; Alam, J. Identification of activating transcription factor 4 (ATF4) as an Nrf2-interacting protein. Implication for heme oxygenase-1 gene regulation. J. Biol. Chem. 2001, 276, 20858–20865.
- Venugopal, R.; Jaiswal, A.K. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc. Natl. Acad. Sci. USA 1996, 93, 14960–14965.
- Venugopal, R.; Jaiswal, A.K. Nrf2 and Nrf1 in association with Jun proteins regulate antioxidant response element-mediated expression and coordinated induction of genes encoding detoxifying enzymes. Oncogene 1998, 17, 3145–3156.
- Katoh, Y.; Itoh, K.; Yoshida, E.; Miyagishi, M.; Fukamizu, A.; Yamamoto, M. Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells 2001, 6, 857–868.
- Ogryzko, V.V.; Schiltz, R.L.; Russanova, V.; Howard, B.H.; Nakatani, Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 1996, 87, 953–959.
- Brown, S.L.; Sekhar, K.R.; Rachakonda, G.; Sasi, S.; Freeman, M.L. Activating transcription factor 3 is a novel repressor of the nuclear factor erythroid-derived 2-related factor 2 (Nrf2)-regulated stress pathway. Cancer Res. 2008, 68, 364–368.
- Shan, Y.; Akram, A.; Amatullah, H.; Zhou, D.Y.; Gali, P.L.; Maron-Gutierrez, T.; Gonzalez-Lopez, A.; Zhou, L.; Rocco, P.R.; Hwang, D.; et al. ATF3 protects pulmonary resident cells from acute and ventilator-induced lung injury by preventing Nrf2 degradation. Antioxid. Redox Signal. 2015, 22, 651–668.
- Ki, S.H.; Cho, I.J.; Choi, D.W.; Kim, S.G. Glucocorticoid receptor (GR)-associated SMRT binding to C/EBPbeta TAD and Nrf2 Neh4/5: Role of SMRT recruited to GR in GSTA2 gene repression. Mol. Cell. Biol. 2005, 25, 4150–4165.
- Zhang, J.; Ohta, T.; Maruyama, A.; Hosoya, T.; Nishikawa, K.; Maher, J.M.; Shibahara, S.; Itoh, K.; Yamamoto, M. BRG1 interacts with Nrf2 to selectively mediate HO-1 induction in response to oxidative stress. Mol. Cell. Biol. 2006, 26, 7942–7952.
- Nioi, P.; Nguyen, T.; Sherratt, P.J.; Pickett, C.B. The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol. Cell. Biol. 2005, 25, 10895–10906.
- Kim, J.H.; Yu, S.; Chen, J.D.; Kong, A.N. The nuclear cofactor RAC3/AIB1/SRC-3 enhances Nrf2 signaling by interacting with transactivation domains. Oncogene 2013, 32, 514–527.
- Pan, H.; Guan, D.; Liu, X.; Li, J.; Wang, L.; Wu, J.; Zhou, J.; Zhang, W.; Ren, R.; Zhang, W.; et al. SIRT6 safeguards human mesenchymal stem cells from oxidative stress by coactivating NRF2. Cell Res. 2016, 26, 190–205.
- Sekine, H.; Okazaki, K.; Ota, N.; Shima, H.; Katoh, Y.; Suzuki, N.; Igarashi, K.; Ito, M.; Motohashi, H.; Yamamoto, M. The Mediator Subunit MED16 Transduces NRF2-Activating Signals into Antioxidant Gene Expression. Mol. Cell. Biol. 2016, 36, 407–420.
- Ikeda, Y.; Sugawara, A.; Taniyama, Y.; Uruno, A.; Igarashi, K.; Arima, S.; Ito, S.; Takeuchi, K. Suppression of rat thromboxane synthase gene transcription by peroxisome proliferator-activated receptor gamma in macrophages via an interaction with NRF2. J. Biol. Chem. 2000, 275, 33142–33150.
- Ansell, P.J.; Lo, S.C.; Newton, L.G.; Espinosa-Nicholas, C.; Zhang, D.D.; Liu, J.H.; Hannink, M.; Lubahn, D.B. Repression of cancer protective genes by 17beta-estradiol: Ligand-dependent interaction between human Nrf2 and estrogen receptor alpha. Mol. Cell. Endocrinol. 2005, 243, 27–34.
- Clarke, R.; Cook, K.L.; Hu, R.; Facey, C.O.; Tavassoly, I.; Schwartz, J.L.; Baumann, W.T.; Tyson, J.J.; Xuan, J.; Wang, Y.; et al. Endoplasmic reticulum stress, the unfolded protein response, autophagy, and the integrated regulation of breast cancer cell fate. Cancer Res. 2012, 72, 1321–1331.
- Pajares, M.; Jimenez-Moreno, N.; Garcia-Yague, A.J.; Escoll, M.; De Ceballos, M.L.; Van Leuven, F.; Rabano, A.; Yamamoto, M.; Rojo, A.I.; Cuadrado, A. Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 2016, 12, 1902–1916.
- Jain, A.; Lamark, T.; Sjottem, E.; Larsen, K.B.; Awuh, J.A.; Overvatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591.