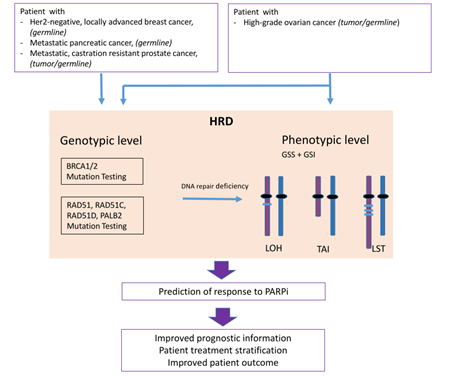
Homologous Recombination Deficiency (HRD) is associated with sensitivity towards PARP inhibitors (PARPi) and its determination is used as a biomarker for therapy decision making.
- DNA double-strand break
- BRCA
- homologous recombination
- HRD score
- BRCAness
- PARPi
1. Introduction
One major underlying mechanism is the emergence of genomic instability caused by genetic mutations arising from exogenously or endogenously caused DNA damage or failures in DNA damage repair. Healthy cells maintain genomic integrity by a variety of repair mechanisms, each addressing unique forms of DNA damage. The base excision repair (BER) pathway is activated by damaged DNA bases (DNA single strand-lesions). In response to double-strand breaks, two different repair pathways are available; the exact mechanism of homologous recombination repair (HRR) and the error-prone non-homologous end joining (NHEJ) [2][1].
Defects in the DNA repair system are an underlying cause of genomic instability due to the accumulation of genetic changes [3][2]. Homologous recombination repair deficiency (HRD) resulting in DNA double-strand breaks is considered to be the most lethal of all DNA repair defects since cancer cells switch to the error-prone NHEJ pathway fostering genomic instability and cell death. PARP inhibition (PARPi) disables single-strand break repair and leads to further accumulation of double-strand breaks, consequently requiring homologous recombination repair. Albeit improved outcome results led to the approval of several inhibtors for different tumor entities, it remains challenging to define those patients who might benefit from PARPi therapy.
2. DNA Repair Mechanisms
Mammalian cells obey different cellular responses to DNA damage, including DNA damage repair, cell cycle arrest and apoptosis. Regarding the responses of DNA repair, different mechanisms can take place. The simplest way of DNA repair is proofreading or direct reversal repair, occurring during replication. The methyl group is transferred to a catalytically active cysteine and afterwards MGMT is targeted to the proteasome for degradation [4][3].Nevertheless, it requires more sophisticated repair mechanisms to effectively correct complex damage on already replicated DNA caused by several endogenous or exogenous factors.
Depending on the factor that causes DNA damage, different types of mismatches/errors occur on single-stranded DNA. For example, radiation modifies single bases or DNA adducts alter the conformation of the DNA. Mismatches created during the replication process due to slippage of DNA strands are corrected by the so-called mismatch repair. Mismatched bases or reading frame shifts are recognised by specific protein dimers, consisting of MSH2 and MSH3 or MSH6.
DNA damage of single bases caused by oxidation, deamination or alkylation are commonly repaired by base excision repair (BER). In a first step, the damaged base is recognised and removed by specialized DNA glycosylases. At this point, the enzyme called Poly (ADP-ribose) polymerase (PARP) binds to the gapped DNA strand and engages proteins required for strand protection and repair. Following this, the DNA strand is sealed by ligases in complex with X-ray repair cross-completing protein 1 (XRCC1) in the short-patch BER.
Two major repair mechanisms have evolved to deal with double-strand lesions, homologous recombination repair (HRR) and non-homologous end joining (NHEJ). The nature of DSBs caused by internal factors, such as replication block, and external influences, such as ionizing radiation, is obviously different. On one hand, lesions associated with the replication process can be directly repaired by using the sister chromatid, which is located in close proximity, as a template for homologous recombination (HR). In order to deal with such types of lesions on DNA in condensed chromatin structures, vertebrates frequently use NHEJ to simply re-ligate the DSB end strands.
Homologous recombination repair (HRR) is an accurate repair mechanism to cope with DSBs. In a first step, the lesion is recognised by the MRE11-RAD50-NBS1 (MRN) complex, which activates ATM (Ataxia telangiectasia mutated) kinase. Upon DNA 5´-end resection, the replication protein (RPA) coats the single-strand DNA regions and activates ATR (Ataxia telangiectasia and Rad3-related) kinase. In the following, RPA is replaced by RAD51 with the help of further repair-associated proteins, such as CHEK2, BRCA1, BRCA2 and PALB2, which are loaded with RAD51. Subsequently, the defective DNA strand attaches to its sister chromatid, which is used as a template for DNA resynthesis. This mechanism of DSB repair is restricted to late S- and G2 phases of the cell cycle [9,10]
The second pathway that evolved to cope with DSBs belongs to the simplest mechanisms: non-homologous end joining (NHEJ). The core component of this pathway is the Ku70–Ku80 complex, which is able to bind ends of double-strand broken DNA and recruit DNA-PKcs to initiate NHEJ. In a final step, DNA ligase IV (LIG4) is simultaneously recruited with XRCC4 and an XRCC4-like factor (XLF) to ligate the processed DNA and restore genome integrity. Thereby, chromosomal integrity is prone to get lost, giving rise to chromosomal rearrangements [2,11][1][4].
The impairment of HRR activity is called homologous recombination repair deficiency (HRD) and is caused by different factors. Additionally, expression of the HRR associated proteins can be diminished by promoter methylation, for example [12][5]. In order to deal with this impairment, cells tend to activate the alternative NHEJ pathway for DSB repair. As already mentioned, this pathway is prone to ending in chromosomal rearrangements.
The most common pathogenic alterations of HRR associated genes occur within the genes BRCA1and BRCA2[14][6]. Nevertheless, other genes of the pathway and their associated proteins can be affected as well. This phenotype of homologous recombination repair deficiency independent of BRCA1/2 mutations is referred to as BRCAness [10][7].
2. PARP Inhibition and Homologous Recombination Repair Deficiency
2.1. The Concept of Synthetic Lethality
2.2. Biomarkers for PARPi: Genes Involved in the Homologous Recombination Repair Pathway
2.3. Biomarker for PARPi: Genomic Instability
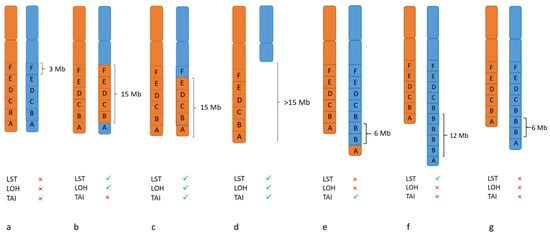
LOH- TAI- LST
References
- Beggs, R.; Yang, E.S. Targeting DNA repair in precision medicine. Adv. Protein Chem. Struct. Biol. 2019, 115, 135–155.
- Jeggo, P.A.; Pearl, L.H.; Carr, A.M. DNA repair, genome stability and cancer: A historical perspective. Nat. Rev. Cancer 2016, 16, 35–42.
- Soll, J.M.; Sobol, R.W.; Mosammaparast, N. Regulation of DNA Alkylation Damage Repair: Lessons and Therapeutic Opportunities. Trends Biochem. Sci. 2017, 42, 206–218.
- De Almeida, L.C.; Calil, F.A.; Machado-Neto, J.A.; Costa-Lotufo, L.V. DNA damaging agents and DNA repair: From carcinogenesis to cancer therapy. Cancer Genet. 2021, 252, 6–24.
- Brianese, R.C.; Nakamura, K.D.M.; Almeida, F.; Ramalho, R.F.; Barros, B.D.F.; Ferreira, E.N.E.; Formiga, M.; de Andrade, V.P.; de Lima, V.C.C.; Carraro, D.M. BRCA1 deficiency is a recurrent event in early-onset triple-negative breast cancer: A comprehensive analysis of germline mutations and somatic promoter methylation. Breast Cancer Res. Treat. 2018, 167, 803–814.
- Heeke, A.L.; Pishvaian, M.J.; Lynce, F.; Xiu, J.; Brody, J.R.; Chen, W.J.; Baker, T.M.; Marshall, J.L.; Isaacs, C. Prevalence of Homologous Recombination-Related Gene Mutations Across Multiple Cancer Types. JCO Precis. Oncol. 2018, 2, 1–13.
- Walsh, C.S. Two decades beyond BRCA1/2: Homologous recombination, hereditary cancer risk and a target for ovarian cancer therapy. Gynecol. Oncol. 2015, 137, 343–350.
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428.
- Hussain, M.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Survival with Olaparib in Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 383, 2345–2357.
- Pommier, Y.; O’Connor, M.J.; de Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362ps317.
- Hodgson, D.R.; Dougherty, B.A.; Lai, Z.; Fielding, A.; Grinsted, L.; Spencer, S.; O’Connor, M.J.; Ho, T.W.; Robertson, J.D.; Lanchbury, J.S.; et al. Candidate biomarkers of PARP inhibitor sensitivity in ovarian cancer beyond the BRCA genes. Br. J. Cancer 2018, 119, 1401–1409.
- Sunada, S.; Nakanishi, A.; Miki, Y. Crosstalk of DNA double-strand break repair pathways in poly(ADP-ribose) polymerase inhibitor treatment of breast cancer susceptibility gene 1/2-mutated cancer. Cancer Sci. 2018, 109, 893–899.
- Ashworth, A. A synthetic lethal therapeutic approach: Poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 3785–3790.
- Miller, R.E.; Leary, A.; Scott, C.L.; Serra, V.; Lord, C.J.; Bowtell, D.; Chang, D.K.; Garsed, D.W.; Jonkers, J.; Ledermann, J.A.; et al. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2020, 31, 1606–1622.
- Lheureux, S.; Lai, Z.; Dougherty, B.A.; Runswick, S.; Hodgson, D.R.; Timms, K.M.; Lanchbury, J.S.; Kaye, S.; Gourley, C.; Bowtell, D.; et al. Long-Term Responders on Olaparib Maintenance in High-Grade Serous Ovarian Cancer: Clinical and Molecular Characterization. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 4086–4094.
- Kondrashova, O.; Topp, M.; Nesic, K.; Lieschke, E.; Ho, G.-Y.; Harrell, M.I.; Zapparoli, G.V.; Hadley, A.; Holian, R.; Boehm, E.; et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat. Commun. 2018, 9, 3970.
- Tung, N.M.; Robson, M.E.; Ventz, S.; Santa-Maria, C.A.; Nanda, R.; Marcom, P.K.; Shah, P.D.; Ballinger, T.J.; Yang, E.S.; Vinayak, S.; et al. TBCRC 048: Phase II Study of Olaparib for Metastatic Breast Cancer and Mutations in Homologous Recombination-Related Genes. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 4274–4282.
- Armstrong, S.A.; Schultz, C.W.; Azimi-Sadjadi, A.; Brody, J.R.; Pishvaian, M.J. ATM Dysfunction in Pancreatic Adenocarcinoma and Associated Therapeutic Implications. Mol. Cancer Ther. 2019, 18, 1899–1908.
- Javle, M.; Shacham-Shmueli, E.; Xiao, L.; Varadhachary, G.; Halpern, N.; Fogelman, D.; Boursi, B.; Uruba, S.; Margalit, O.; Wolff, R.A.; et al. Olaparib Monotherapy for Previously Treated Pancreatic Cancer with DNA Damage Repair Genetic Alterations Other Than Germline BRCA Variants: Findings from 2 Phase 2 Nonrandomized Clinical Trials. JAMA Oncol. 2021, 7, 693–699.
- Caracciolo, D.; Riillo, C.; Di Martino, M.T.; Tagliaferri, P.; Tassone, P. Alternative Non-Homologous End-Joining: Error-Prone DNA Repair as Cancer’s Achilles’ Heel. Cancers 2021, 13, 1392.
- Plon, S.E.; Eccles, D.M.; Easton, D.; Foulkes, W.D.; Genuardi, M.; Greenblatt, M.S.; Hogervorst, F.B.; Hoogerbrugge, N.; Spurdle, A.B.; Tavtigian, S.V. Sequence variant classification and reporting: Recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum. Mutat. 2008, 29, 1282–1291.
- Cline, M.S.; Liao, R.G.; Parsons, M.T.; Paten, B.; Alquaddoomi, F.; Antoniou, A.; Baxter, S.; Brody, L.; Cook-Deegan, R.; Coffin, A.; et al. BRCA Challenge: BRCA Exchange as a global resource for variants in BRCA1 and BRCA2. PLoS Genet. 2018, 14, e1007752.
- Hauke, J.; Horvath, J.; Groß, E.; Gehrig, A.; Honisch, E.; Hackmann, K.; Schmidt, G.; Arnold, N.; Faust, U.; Sutter, C.; et al. Gene panel testing of 5589 BRCA1/2-negative index patients with breast cancer in a routine diagnostic setting: Results of the German Consortium for Hereditary Breast and Ovarian Cancer. Cancer Med. 2018, 7, 1349–1358.
- Hoppe, M.M.; Sundar, R.; Tan, D.S.P.; Jeyasekharan, A.D. Biomarkers for Homologous Recombination Deficiency in Cancer. J. Natl. Cancer Inst. 2018, 110, 704–713.
- Watkins, J.A.; Irshad, S.; Grigoriadis, A.; Tutt, A.N. Genomic scars as biomarkers of homologous recombination deficiency and drug response in breast and ovarian cancers. Breast Cancer Res. BCR 2014, 16, 211.
- Abkevich, V.; Timms, K.M.; Hennessy, B.T.; Potter, J.; Carey, M.S.; Meyer, L.A.; Smith-McCune, K.; Broaddus, R.; Lu, K.H.; Chen, J.; et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer 2012, 107, 1776–1782.
- Birkbak, N.J.; Wang, Z.C.; Kim, J.Y.; Eklund, A.C.; Li, Q.; Tian, R.; Bowman-Colin, C.; Li, Y.; Greene-Colozzi, A.; Iglehart, J.D.; et al. Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov. 2012, 2, 366–375.
- Popova, T.; Manié, E.; Rieunier, G.; Caux-Moncoutier, V.; Tirapo, C.; Dubois, T.; Delattre, O.; Sigal-Zafrani, B.; Bollet, M.; Longy, M.; et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012, 72, 5454–5462.
- Timms, K.M.; Abkevich, V.; Hughes, E.; Neff, C.; Reid, J.; Morris, B.; Kalva, S.; Potter, J.; Tran, T.V.; Chen, J.; et al. Association of BRCA1/2defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 2014, 16, 475.
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallasi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 3764–3773.
- Fuh, K.; Mullen, M.; Blachut, B.; Stover, E.; Konstantinopoulos, P.; Liu, J.; Matulonis, U.; Khabele, D.; Mosammaparast, N.; Vindigni, A. Homologous recombination deficiency real-time clinical assays, ready or not? Gynecol. Oncol. 2020, 159, 877–886.
- Stover, E.H.; Fuh, K.; Konstantinopoulos, P.A.; Matulonis, U.A.; Liu, J.F. Clinical assays for assessment of homologous recombination DNA repair deficiency. Gynecol. Oncol. 2020, 159, 887–898.