Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Patrick Ming Kuen Tang and Version 2 by Conner Chen.
Transforming growth factor-β (TGF-β) signaling triggers diverse biological actions in inflammatory diseases. In tissue fibrosis, it acts as a key pathogenic regulator for promoting immunoregulation via controlling the activation, proliferation, and apoptosis of immunocytes. In cancer, it plays a critical role in tumor microenvironment (TME) for accelerating invasion, metastasis, angiogenesis, and immunosuppression. Increasing evidence suggest a pleiotropic nature of TGF-β signaling as a critical pathway for generating fibrotic TME, which contains numerous cancer-associated fibroblasts, extracellular matrix proteins, and remodeling enzymes. Better understanding the underlying mechanisms may uncover novel therapeutic targets for cancer.
- TGF-β
- tumor microenvironment
- fibrosis
- cancer
- Smad
1. TGF-β Signaling
The TGF-β superfamily was discovered as a group of cytokines with shared properties in synthesis, signal transduction mechanisms, and functions [1][2][14,15]. Eventually, three isoforms, TGF-β1, TGF-β2, and TGF-β3, were eventually identified, synthesized as inactive precursor forms that need to be further activated for triggering their downstream canonical or non-canonical pathways [3][7]. C-terminal TGF-β homodimer, latency-associated peptide (LAP), and N-terminal signal peptide are three components of the TGF-β precursor. Cleavage of this precursor removes the signal peptide, leaving the TGF-β homodimer and LAP together, known as the small latent complex (SLC) [3][4][7,11]. Then, the SLC form a disulfide linkage to the latent TGF-β binding protein (LTBP), leading to the production of a large latent complex (LLC), which is often associated with ECM and remains inactive [3][7]. The LLC can be further cleaved by proteases to release the active TGF-β homodimer for activating its receptors to initiate the downstream signaling.
1.1. Canonical Pathway
Conventionally, the active TGF-β binds to the TGF-β receptor type II, which recruits and phosphorylates type TGF-β receptor type I (TGF-βR1) [4][11]. The activated TGF-βR1 will phosphorylate the C-terminal serine residues of the receptor-associated Smads (R-Smads) Smad2 and Smad3, then, the R-Smads will dissociate from type I receptor and form a heterotrimeric complex with common Smad (Co-Smad) Smad4 at the early endosome and translocate into the nucleus [1][5][14,16]. The Smad complexes will interact with transcription factors, chromatin binding proteins and transcription co-activators and co-repressors in the nucleus and physically bind to the target genes for executing the transcriptional regulation at genomic level [6][5][8,16], resulting in the diverse effects of the TGF-β canonical pathway in different contexts. There is also another Smad member called inhibitory Smad (I-Smad), e.g., the classic example Smad7, which negatively regulates the canonical pathway by competing with Smad 2/3 for binding of type I receptor as a negative feedback for attenuating the TGF-β/Smad signaling [4][11], as summarized in Figure 1.
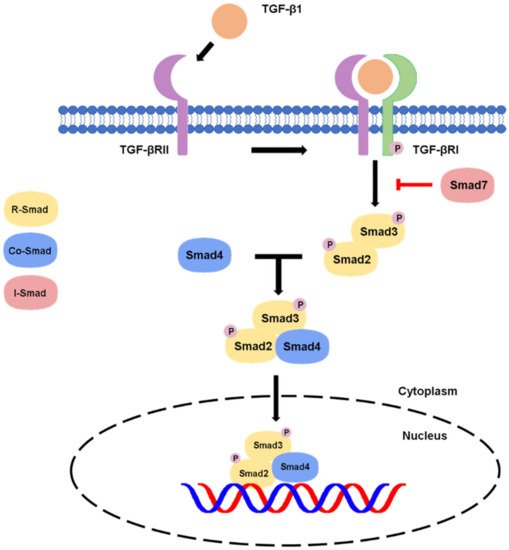
Figure 1. Canonical pathway of TGF-β signaling.
In canonical signaling cascade, the activated TGF-βR1 leads to Smad2/3 phosphorylation. The Smad2/3 complex then binds to Smad4 and translocates into the nucleus, where it induces gene transcription. The signaling can be inhibited by a negative feedback of Smad7 on the TGF-βR1.
1.2. Non-Canonical Pathway
Besides, there are non-canonical pathways of the TGF-β1 signaling, where extracellular signal-regulated kinase (Erk)/mitogen-activated protein kinase (MAPK) signaling pathway are involved in the downstream signal transductions. After the phosphorylation of TGF-β type I and II receptors at tyrosine residues, adaptor proteins Src homology 2 (SH2) domain-containing protein A (ShcA) and growth factor receptor-bound protein 2 (Grb2)/son of sevenless (SOS) complex is recruited [7][17]. The Grb2/SOS complex catalyzes the exchange of GDP to GTP for activating a small GTPase Ras, which then triggers the gene regulatory actions via Raf, MEK1/2, and Erk1/2 [7][8][17,18]. For example, Erk1/2 can phosphorylate targeted transcription factors such as Fos-related antigen 2 (Fra-2) in order to promote gene transcription [8][9][18,19]. In addition, protein kinase B (Akt) can be activated by TGF-β signaling via phosphatidylinositol-3 kinase (PI3K) for regulating translational responses though mTOR [7][17]. Akt can also be activated in the non-canonical pathway via TRAF6-mediated Akt lysine-63 chain ubiquitination or Smad7 phosphatase and tensin homolog (PTEN) inhibition by miR-216a/217 microRNA cluster [10][11][20,21]. Moreover, RhoA and Rho-associated protein kinase (ROCK) can also been regulated by TGF-β signaling through a Smad independent manner [7][17]. In addition, TGF-β mediates epithelial-mesenchymal transformation (EMT) of cancer cells by interfering with cell adhesion and epithelial gene expression, as well as by increasing the expression of mesenchymal proteins such as fibronectin, N-cadherin, vimentin, and fibronectin [12][22]. Activation of the Smad-dependent pathway induces the expression of SNAIL, SLUG, ZEB, and TWIST transcription factors, which act as E-cadherin repressors and mediate the dissociation of desmosomes [13][23]. On the other hand, activation of Smad-independent pathway promotes cytoskeletal remodelling by ERK activation and tight junction dissolution via Rho GTPase [14][24]. ERK strengthens the transcriptional activity of Smad, thereby assisting TGF-β/Smad dependent EMT [15][25]. The non-canonical pathways are systemically shown in Figure 2.
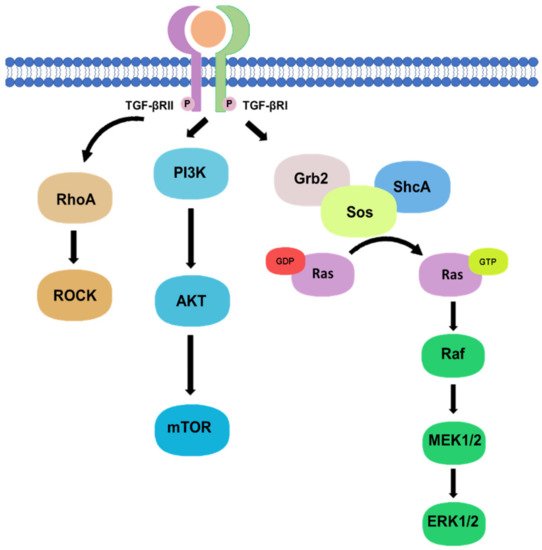
Figure 2. Non-canonical pathway of TGF-β signaling.
Phosphorylation of TGF-β type I and II receptors can also activate downstream non-canonical pathway including Rho, PI3K/Akt, and Grb2/SOS signaling in a Smad-independent manner.
2. TGF-β Signaling in Tissue Fibrosis
Tissue fibrosis is the major pathological feature of most chronic inflammatory diseases, which results in the failure of important organs and can lead to mortality [16][26]. In chronic kidney disease (CKD), fibrosis is an essential step in the development of end-stage renal disease (ESRD) [17][18][19][20][21][27,28,29,30,31], where TGF-β/Smad-dependent signaling pathway plays a critical role [22][23][4,32]. TGF-β1 medicates ECM synthesis and degradation in progressive renal fibrosis. TGF-β1 also induces the transformation of tubular epithelial cells to myofibroblasts through EMT to cause renal fibrosis [23][24][32,33]. The primary downstream mediators of TGF-β1, Smad2, and Smad3, are extensively activated in fibrotic kidneys in patients and animal models with CKD [25][34]. As fibrosis progresses, Smad2 is protective, while Smad3 leads to pathogenic changes. Smad3 directly binds to the promoter region of collagens to trigger renal fibrosis production and reduces the activity of MMP-1 to inhibit ECM degradation via induction of TIMP-1 [26][27][28][29][35,36,37,38]. By contrast, conditional knockout of Smad2 from tubular epithelial cells enhances Smad3-mediated renal fibrosis, which is associated with phosphorylation and nuclear translocation of Smad3, auto-induction of TGF-β1 expression, and transcription of collagen I and III genes [30][39]. In response to TGF- β1 and BMPs, Smad4 and Co-Smad promote nuclear translocation of Smad2/3 and Smad1/5/8 complexes, respectively [25][31][34,40]. Deficiency of Smad4 in mesangial cells in vitro dramatically suppresses collagen I promoter activity and significantly reduces fibrosis in the mice tubular epithelial cells with UUO-induced fibrosis without affecting Smad3 activation [32][41]. Smad7 is the inhibitor of TGF/Smad canonical signaling in fibrosis. Smad7 can compete with Smad2 and Smad3 for TGF-βR1 activation as well as ubiquitinated and degraded Smad2 and TGF-βRI by recruiting the E3 ubiquitin ligase Smad ubiquitination regulatory factors [33][34][35][42,43,44].
Extensive studies demonstrated the crucial role of TGF-β1 signaling in the pathogenesis of liver diseases, including hepatitis and cirrhosis as well as hepatocellular carcinoma [36][45]. In hepatic fibrosis, type I collagen expression and epithelial-myofibroblast transition are actively produced by up-regulating the pro-fibrotic SMAD3 but down-regulating the anti-fibrotic SMAD2. The responsive promoter activity of SMAD3 can be enhanced by SMAD4 but blocked by its native negative mediator SMAD7 [37][46].
Heart failure is an increasingly prevalent disease in humans, a progressive loss of cardiomyocytes, ventricular chamber remodeling, and accumulation of interstitial fibrosis resulting in a lethal reduction of cardiac output [38][47]. Several studies have explored the potential contributions of both canonical and non-canonical pathways of the TGF-β signaling in the pathogenesis of cardiac fibrosis [39][40][41][48,49,50]. In addition, idiopathic pulmonary fibrosis (IPF) and interstitial lung fibrosis are particularly austere lung diseases [42][51], where TGF-β signaling is one of the most potent profibrotic inducer for accelerating the progression of lung fibrosis through recruiting and activating monocytes and fibroblasts as well as induction of ECM production in the lesion [43][52] (Figure 3). Importantly, increasing evidence revealed the importance of crosstalk between TGF-β and other signaling pathways in the pathogenesis of tissue fibrosis. For example, TGF-β1 increases the MMP production of lung stromal fibroblast through Wnt/β-catenin signaling pathway to future enhance the development of IFP [44][53]. Moreover, TGF-β signaling also works with a Hippo-YAP/TAZ pathway for modulating cardio fibrosis via up-regulating the production of TGF-β1 [45][54].
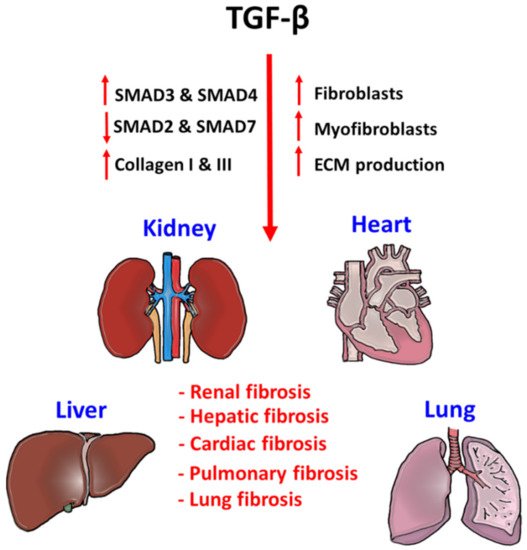
Figure 3. Pathogenic role of TGF-β signaling in tissue fibrosis.
TGF-β signaling is a critical factor for initiating fibrogenesis in kidney, liver, heart, and lung. Phosphorylation of pro-fibrotic SMAD3 and SMAD4, and repression of anti-fibrotic SMAD2 and SMAD7 largely enhance the fibroblast proliferation, myofibroblast differentiation, and ECM production in the injured tissues. Accumulation of tissue stiffness and scar tissue largely affects their physiological functions and lead to organ failure.
3. TGF-β Signaling in the Tumor Microenvironment
Increasing evidence demonstrated an equal weight of both the adaptive and innate immunity in the cancer progression, which can be largely suppressed by cancer cells via TGF-β1 signaling. In adaptive immunity, elevated TGF-β-levels suppress T cell development and anticancer function [46][55]. TGF-β blocks Th1 and CD8+ cytotoxic T cells differentiation from naïve T cell by inhibiting the expression of T-box transcription factor (T-bet) at a transcriptional level [47][48][56,57]. TGF-β also supresses T cells proliferation via diminishing the expression of Interleukin-2 (IL-2) by promoting the interaction of Smad4 on an anti-proliferative mediator TOB at a genomic level [49][58]. In fact, early detection of SMAD4 in pancreas help stratify pancreatic cancer patients for new therapy selection [50][59]. Importantly, the regulatory role of TGF-β1 signaling has been emergingly observed in the innate immunity. Tumor-associated macrophage (TAM) can be further polarised to show pro-tumoral M2 phenotypes in the TME for exhibiting pro-angiogenic, anti-inflammatory, and immunosuppressive features to promote tumor development [51][60]. Recent evidence suggested that TGF-β can trigger the M1/M2 polarization of TAMs via activating the Smad2/3 and PI3K/AKT pathways for enhancing the transcription of pro-tumoral effectors, including IL-10, VEGFA and CXCR4 [52][61]. Moreover, TGF-β1/Smad3 signaling diminish the production of an anti-tumor cytokine, interferon-γ (IFN-γ) in natural killer cells (NK cells) by T-bet suppression to inhibit their cytotoxic function [53][62]. In addition, TGF-β1/Smad3 signaling can also markedly suppress the development of NK cells via downregulating the transcription factor E4BP4 via transcriptional regulation in a T-bet independent manner [54][9].
TGF-β is also crucial for activating cancer-associated fibroblasts (CAFs) to modulate migration and invasion of the cancer cells [55][56][63,64]. TGF-β is able to induce the transition process of fibroblasts towards myofibroblasts by increasing fibroblast contractility and CAF marker expression including α-smooth muscle actin (α-SMA), fibroblast activation protein (FAP), tenascin-C and platelet-derived growth factor receptor (PDGFR) [57][58][59][60][65,66,67,68]. Increasing evidence suggests that TGF-β1 can enhance the expression of ECM remodelling gene ACTA2 (α-SMA) [61][62][69,70], collagen precursor PLOD2, and membrane bound thymocyte differentiation antigen Thy1/CD90 for supporting the CAF formation [63][71]. Furthermore, angiogenesis is one of the critical steps in tumorigenesis, where TGF-β/Smads signaling is significantly involved in the regulatory machinery, such as vascular endothelial growth factor (VEGF), fibroblast growth factor-1 (FGF-1), and platelet-derived growth factor (PDGF) [64][65][72,73]. TGF-β also interacts with the type I receptors, activin receptor–like kinases 1 (ALK1) or 5 (ALK5) to regulate angiogenesis, where TGF-β/ALK1 signaling induces downstream signal via Smad1/5 while TGF-β/ALK5 uses Smad2/3 to regulate the angiogenic factors, respectively [65][66][73,74]. Activation of ALK1 requires activation of ALK5, as the lack of TGF-β/ALK5 activation will deteriorate both pathways, highlighting TGF-β as the key regulator to balance ALK1 and ALK5 in endothelial cells for mediating angiogenesis in cancer [65][73]. Indeed, TGF-β/Smad pathway also mediates endothelial-mesenchymal transformation via SNAIL/Slug expression in endothelial cells (TECs) to support sprouting angiogenesis, and accumulation of myofibroblast and CAFs in the TME [67][10] (Figure 4).
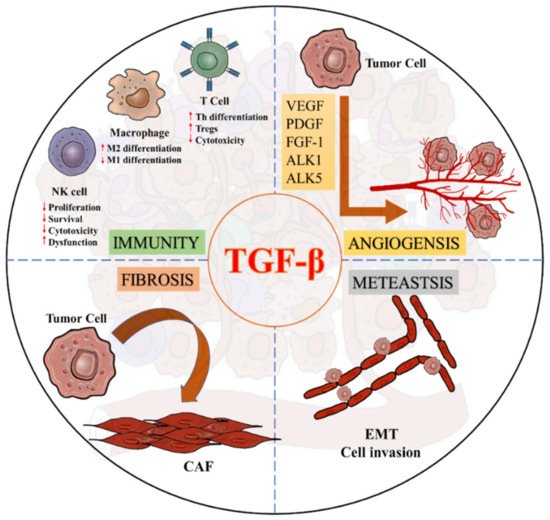
Figure 4. Diverse roles of TGF-β signaling in a pro-tumoral TME.