Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Federica Cherchi and Version 3 by Camila Xu.
Oligodendrocytes (OLs) are ramified glial cells within the central nervous system (CNS) whose terminal processes generate myelin and enwrap neuronal axons.
- oligodendrocyte precursor cells
- oligodendrocyte differentiation
- voltage-gated ion channels
- neurotransmitter receptors
1. Introduction
Oligodendrocytes (OLs) are ramified glial cells within the central nervous system (CNS) whose terminal processes generate myelin and enwrap neuronal axons. Myelin is crucial for the saltatory propagation of electrical impulses along nerve fibres, enabling rapid communication between networks in the CNS [1]. In addition to myelin deposition, OLs secrete metabolic factors and maintain energy homeostasis to support axonal integrity and promote neuronal survival [2].
Thus, a deeper understanding of OL functions during brain development, as well as during their regeneration in neurological disorders that involve OL and myelin loss, is crucial to understand their homeostatic functions within the CNS function and to identify new therapeutic targets for demyelinating diseases.
During CNS development, OL progenitor cells (OPCs) are generated from neural stem/progenitor cells (NSPCs) in several regions in a precise spatiotemporal manner [3][4][3,4]. Multiple transcriptional regulators cooperate to orchestrate changes in gene expression leading to OPC fate selection and subsequent differentiation into OL. Details on intrinsic signals regulating oligodendroglial cell specification and progression have been described [5][6][5,6]. However, since OLs are part of a complex environment containing neurons, astrocytes, microglia, and vascular/perivascular cells, the control of oligodendrogliogenesis likely relies on multiple extrinsic cues and cell–cell interactions during development or regeneration.
2. Oligodendrogliogenesis
Only 5–8% of total glial cells are OPC, which are evenly distributed in white and grey matter [7] with different functions; OPCs in white matter present a higher proliferative response to platelet-derived growth factor (PDGF)-A and enhanced differentiation into myelinating OLs than OPC in grey matter [8].
Oligodendrogliogenesis and remyelination are crucial events for white matter reorganization [9]. Among other functions, OLs support and regulate axonal electrical activity by producing myelin not only to increase action potential (AP) conduction, but also to provide metabolic support and stabilize axonal cytoskeleton [10][11][10,11]. After acute traumatic injury in rodents, there is an active proliferation of OPCs derived from NSPCs in the subventricular zone [12]. Migration of oligodendroglial cells from the proliferative zones to their final position is an essential step during CNS development and myelination. In humans, increased OPC pool at sites of ischemic brain insults has been observed post-mortem. However, only a fraction of proliferating OPCs become mature functional OLs and contribute to remyelination. Furthermore, there is an age-related decline in normal oligodendrogliogenesis. Interestingly, a recent study demonstrated the possibility of exercise to increase OPCs in white matter as a potent therapy for neural circuit remodelling [13].
In order to study oligodendrogliogenesis, several markers are available to identify different steps of OPC maturation (Figure 1). Once committed to the oligodendroglial lineage, cell surface antigens can be recognized by specific antibodies, such as A2B5 [14]. Moreover, SRY-Box Transcription Factor 9 (Sox9) is also strongly expressed first in NSPCs, and later in glial cells of the CNS, and is essential for proper development of both OLs and astrocytes; Sox9 continues to be expressed after specification into OPCs [15].
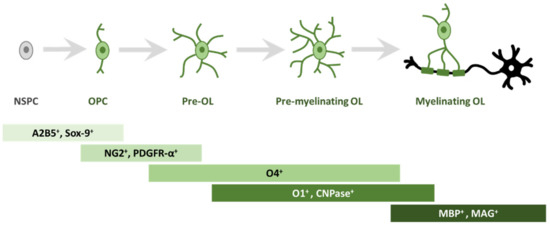
Figure 1. Schematic representation of morphological and antigen expression changes during oligodendrogliogenesis. A specific array of antigen expression and cell morphology has been associated to different steps of oligodendrogliogenesis, from oligodendrocyte precursor cells (OPC) to mature myelinating oligodendrocyte (OL). Abbreviations: cell surface ganglioside epitope (A2B5); SRY-Box Transcription Factor 9 (Sox9); neuron-glial antigen 2 (NG2) proteoglycan; receptor for PDGF-A (PDGFR-α); cell surface markers (O4); cell surface markers (O1); 2′, 3′-cyclic-nucleotide 3′-phosphodiesterase (CNPase); myelin basic protein (MBP); myelin associated glycoprotein (MAG).
Among the best known OPC markers are PDGFR-α, the receptor for PDGF-A, and the neuron-glial antigen 2 (NG2) proteoglycan [16][17][18][16,17,18]. PDGF-A is the most potent OPC mitogen and survival factor, which is produced by both astrocytes and neurons; consequently, overexpression of this growth factor, e.g., during development, leads to increased OPC number [19]. When pre-OLs engage with a target axon, they lose bipolarity to acquire a ramified morphology and start to build filamentous myelin outgrowths [8]. At this differentiation stage, pre-OLs are characterised by the expression of three main myelin-associated markers, 2′, 3′-cyclic-nucleotide 3′-phosphodiesterase (CNPase) and the cell surface markers O4 and O1 [20]. O4 is already expressed in late progenitors, whereas O1 is typical of pre-myelinating OLs [21]. Mature, differentiated OLs are characterised by the production of myelin and myelin-related proteins, such as myelin basic protein (MBP) [22], which is expressed on the cytoplasmic surface of the plasma membrane [23], the transmembrane protein myelin proteolipid protein (PLP) [24] and myelin associated glycoprotein (MAG) [25].
3. Voltage-Gated Channels in Oligodendroglial Cells and Myelination
OPCs receive synaptic inputs from neurons [26] and express voltage-gated ion channels (such as Na+ and K+ channels; Figure 2) and various neurotransmitter receptors [27][28][29][30][31][27,28,29,30,31]. During motor learning in mice, a rapid differentiation of OPCs in the motor cortex was found, followed by a subsequent increase in compensatory proliferation to return to homeostatic OPC density [32]. Moreover, it was recently demonstrated that motor learning could enhance oligodendrogliogenesis and remyelination after cuprizone (CPZ) intoxication, a mice model of demyelination [33]. Similarly, oligodendrogliogenesis and de novo myelination were increased following spatial learning in the water maze test [34]. Social experienced after cuprizone treatment also influenced remyelination in adult mice [35] and oligodendrogliogenesis in adolescent mice [36]. These studies imply that activation of neuronal circuits plays an important role in white matter development [37]. By speeding AP conduction, myelination increases the brain’s cognitive abilities. During development or learning, a re-arrangement of myelin thickness or internode length may help to tune the speed of conduction along myelinated axons [38]. This can promote synchronous neuronal firing [39], make impulse propagation time less dependent on the spatial trajectory of the axon transmitting information between areas [40], or adjust propagation delays along the cochlear nerve to promote sound localization [41]. Magnetic resonance imaging (MRI) reveals changes in white matter microstructure, perhaps reflecting alterations of myelin, when human subjects learn a skilled motor task such as playing piano [42] or juggling [43]. Together, these studies suggest that adaptive myelination is a physiological and essential aspect of neural plasticity and reveal the presence of an important crosstalk between neurons and OLs.
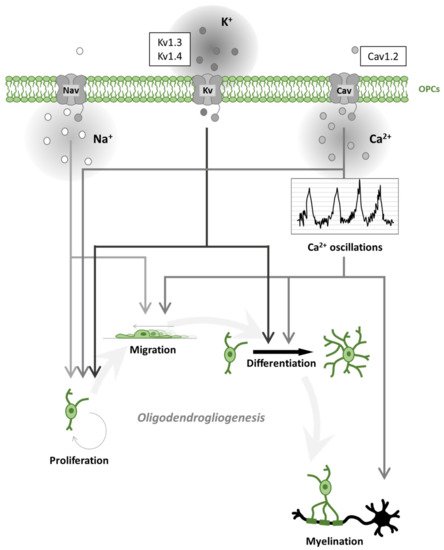
Figure 2. Oligodendrogliogenesis is differentially regulated by voltage-gated ion channels. Schematic representation of voltage-gated Na+, K+, Ca2+ channel (Nav, Kv, Cav) effects on oligodendrogliogenesis. Nav contributes to oligodendrocyte precursor cell (OPC) proliferation and migration. Kv regulates proliferation and differentiation. Cav regulates all steps of oligodendrogliogenesis. In particular, Cav activation contributes to spontaneous Ca2+ oscillations leading to accelerated migration, process formation (differentiation) and myelination. The most expressed ion channels, within each ion-selective group, are represented in the sidebar upwards.
Among demyelinating pathologies, multiple sclerosis (MS) represents the most common chronic disease in the CNS. MS is primarily considered an immune-mediated disease and is characterized by focal areas of infiltrated lymphocytes causing neuroinflammation with associated demyelination that spreads over the brain and spinal cord with time [44]. The classic pathological hallmark of MS was long considered to be the presence of focal white matter demyelinating lesions. However, pathological changes are also detectable in normal-appearing white matter (NAWM), as well as in the CNS grey matter, with the presence of focal grey matter lesions and grey matter atrophy [45]. Indeed, it can be supposed that one of the mechanisms involved in the pathogenesis of MS is the altered gene expression and reorganization of ion channels at the level of demyelinated axons [46].
It is now known that ion channels are considered an important target class for the study and treatment of various pathologies. In particular, the Food and Drug Administration (FDA) has recognized both ligand-dependent and voltage-dependent ion channels among the top pharmaceutical targets of approved agents [47]. Therefore, ion channel dysregulation on neurons and glial cells can probably contribute to axonal degeneration during chronic inflammation in the brain and spinal cord [46][48][46,48] and to abnormal activation in immune cells. These hypotheses are supported by numerous studies where ion channel blockers have been shown to modulate CNS damage and symptoms due to experimental autoimmune encephalomyelitis (EAE), a mouse model for MS. Indeed, different studies on EAE mice revealed distinct ion channel families as key players in pathophysiological processes of demyelination and could be important drug targets for this disease [49][50][49,50].
According to their vast expression, ion channels have the potential to influence nearly every stage of MS pathogenesis. The regulation of the immune response is, among other modulators, dependent on ion channels which allow peripheral T lymphocytes to proliferate and to produce inflammatory cytokines [51].
Within the class of drugs targeting ionic channels on immune cells is Glatiramer Acetate (GA), an immunomodulatory drug used in the treatment of MS, which seems to play an important role on B lymphocytes, where it modifies both the immune response and the activation of ion channels (see Table 1). In the first case, GA inhibits B lymphocytes maturation, with a concomitant increase in the number B cell precursors and/or naïve B cells. In the second case, it was observed that GA was able to modulate Ca2+ homeostasis in these cells. Indeed, it was observed that GA changed the expression of K+ and Cl− channels but, also, Ca2+ release activated Ca2+ entry and transient receptor potential (TRP) channel opening [52].
Table 1.
Voltage-gated ion channel-targeting compounds and their role in demyelinating conditions.
| Drug/s | Ion Channel/s | Preclinical/Clinical Trials | Effects | ||||||
|---|---|---|---|---|---|---|---|---|---|
| PF-01247324 | Nav1.8-selective blocker | EAE | Improves motor coordination and cerebellar-like symptoms [53] | ||||||
| Safinamide | Unselective Nav blocker | EAE | Protects from neurological deficit and prevents microglial activation [54][55] | Protects from neurological deficit and prevents microglial activation [54,55] | |||||
| Flecainide | Nav blocker | EAE | Preserves axonal integrity and electrical conduction, reduces disability scores [56][57] | Preserves axonal integrity and electrical conduction, reduces disability scores [56,57] | |||||
| Phenytoin | Nav blocker | EAE Phase II |
Preserves axonal integrity and electrical conduction, reduces disability scores [56] | Preserves axonal integrity and electrical conduction, reduces disability scores [56 | [57] | ,57] | |||
| Neuroprotective in MS and related optic neuritis demyelination [58][59] | Neuroprotective in MS and related optic neuritis demyelination [58,59] | ||||||||
| Lamotrigine | Nav blocker | EAE Phase II |
Preserves axonal integrity and electrical conduction, reduces disability scores [56][57] | Preserves axonal integrity and electrical conduction, reduces disability scores [56,57] | |||||
| Protective effects in preclinical models but no effect on cerebral volume changes [58][60] | Protective effects in preclinical models but no effect on cerebral volume changes [58,60] | ||||||||
| Side effects: reduction in white matter volume in secondary progressive MS patients [58][60] | : reduction in white matter volume in secondary progressive MS patients [58,60] | ||||||||
| Carbamazepine | Nav blocker | EAE Phase II |
Improves paroximal dysarthria and ataxia in MS patients [61] | ||||||
| Pregabalin | Cav blocker | EAE Phase II |
Reduces neuropathic pain in MS patients or EAE model. | ||||||
| Neuroprotective during excitotoxicity or neuroinflammation in EAE. | |||||||||
| Side effects: reduces long-term potentiation in EAE mice and impairs memory function in MS patients [58][62][63][64][65] | : reduces long-term potentiation in EAE mice and impairs memory function in MS patients [58,62,63,64,65] | ||||||||
| Bepridil, Nitrendipine | L-type Cav1.x blocker | EAE | Reduces neuroinflammation and axonal pathology in EAE [66] | ||||||
| Nimodipine | L-type Cav1.2 blocker | EAE | Reduces EAE severity and demyelination [67] | ||||||
| Antiapoptotic effect by preventing intracellular Ca | 2+ overload [68][69] Anti-inflammatory effect by preventing microglial activation [70][71][72][73] | overload [68,69] Anti-inflammatory effect by preventing microglial activation [70,71,72,73] |
|||||||
| BgK-F6A | Kv1.1 selective blocker | CPZ model EAE |
Enhances remyelination in CPZ model [74] | ||||||
| Reduces EAE severity [75] | |||||||||
| Dalfampridine | I | A | blocker | EAE FDA approved in 2010 |
Enhances axonal conduction in EAE [76] | ||||
| Improves motor activity (walking) in MS patients [77] | |||||||||
| Glatiramer Acetate | Modulate K | + | , Cl | − | , Ca | 2+ | and TRP channels [52] | FDA approved in 1996 | Inhibits B lymphocytes maturation |
Nav: voltage-gated sodium channels; Cav: voltage-gated calcium channels; Kv: voltage-gated potassium channels; IA: transient A-type potassium current; TRP: Transient Receptor Potential; EAE: Experimental Autoimmune Encephalomyelitis; CPZ: cuprizone; FDA: Food and Drug Administration; MS: Multiple Sclerosis.
A bulk of evidence indicates that the application of ion channel blockers, such as those for Na+, Ca2+ and K+, greatly improves EAE disease course and delays the pathology onset after immunization in comparison to sham-treated control mice. Based on above evidence, ion channels, expressed either on central or peripheral cells, are regarded as putative promising new targets for MS, particularly in the progressive form of the disease. Indeed, the beneficial effect of ion channel blockers observed in pre-clinical studies may be mediated by two distinct pathways: modulation of an ion channel on nerve cells, which may facilitate neuronal survival, as well as inhibition of ion channels on T cells, which may provide immunomodulation [78].
Currently, clinical translation of ion channel-targeting compounds remains a major challenge in MS. Indeed, only a few clinical trials have been performed and most approaches are still at early preclinical stage, possibly due to the lack of selective compounds, on one side, and poor knowledge of their exact role in the context of autoimmunity, on the other [78].
3.1. Voltage-Gated Na
+
Channels in Oligodendroglial Cells
As summarized in Table 2, TTX-sensitive voltage-gated Na+ channels (Nav) were found on A2B5+/NG2+ OPC [28][79][80][81][82][83][84][28,79,80,81,82,83,84], but their expression is restricted to the earliest stages of OPC differentiation, being down-regulated during maturation into OLs [85][86][87][85,86,87]. Nav have also been found in the white matter of cortical slices obtained from P3-P8 animals, but not at later ages (P10-P18) [83][86][83,86]. In confirm, Paez and colleagues investigated Na+ currents in oligodendroglial cells recorded from brain slices of transgenic mice expressing green fluorescent protein (GFP) under the control of the PLP gene promoter (OL-GFP) [86][88][86,88]. In these transgenic OL-GFP animals (P4-P6), the majority of O4+ cells (91%) in the corpus callosum, representing immature pre-myelinating OLs, have spherical cell bodies with multiple short radiating processes and lack inward Na+ currents [86][89][86,89]. By contrast, most of Sox9+ cells (90%) close to the lateral ventricles have simple morphology with few processes and expressed abundant Nav [86].
Table 2. Expression and functional role of ion channels in oligodendroglial cells in vitro, in MS in vivo animal models or MS patients.
| Ion Channel | OPC/OL Culture In Vitro | In Vivo MS Animal Models | MS Patients | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nav | Expression | TTX-sensitive Nav expressed in OPC, downregulated in OL [28][79][80][81][82][83][84][85][86][87][88] | TTX-sensitive Nav expressed in OPC, downregulated in OL [28,79,80,81,82,83,84,85,86,87,88] | Nav1.2 and Nav1.6 overexpressed in demyelinated sites (EAE) [53][90] Nav1.8 upregulated in cerebellar Purkinje cells (EAE) [91] | Nav1.2 and Nav1.6 overexpressed in demyelinated sites (EAE) [53,90] Nav1.8 upregulated in cerebellar Purkinje cells (EAE) [91] |
Nav1.8 upregulated in cerebellar Purkinje cells [91] Nav1.5 expressed in reactive astrocytes [92] Nav1.6 colocalizes with amyloid precursor protein in post-mortem tissues from secondary progressive MS brain [93] |
||||||||||||
| Function | ↑ Migration and proliferation [94][95] | ↑ Migration and proliferation [94,95] | ||||||||||||||||
| Cav | Expression | L-type (Cav1.2, Cav1.3) in OPC (downregulated in OL) [81][83][96][97][98] N-type (Cav2.2) in OL [99] | L-type (Cav1.2, Cav1.3) in OPC (downregulated in OL) [81,83,96,97,98] N-type (Cav2.2) in OL [99] |
Cav1.2 involved in remyelination (CPZ) [100][101] Increased activity of L-type in demyelinated corpus callosum (CPZ) [102] Overexpression of N-type (Cav2.2) in active lesions (EAE) [103] | Cav1.2 involved in remyelination (CPZ) [100,101] Increased activity of L-type in demyelinated corpus callosum (CPZ) [102] Overexpression of N-type (Cav2.2) in active lesions (EAE) [103] |
Overexpression of N-type (Cav2.2) in active lesion [103] | ||||||||||||
| Function | L-type: ↑ Migration [104][105][106][107 | L-type: ↑ Migration [104,105 | ] | ,106 | [108] ↑ Maturation | ,107 | [ | ,108 | 96][109]↑ Proliferation | ] ↑ Maturation [96 | [86 | ,109 | ][96 | ]↑ Proliferation [86 | ] | ,96] | ||
| Kir | Expression | Kir4.1 OPC > OL [98][106][110] Kir2.1, Kir7.1 and TASK1 in OL [111][112][113] | Kir4.1 OPC > OL [98,106,110] Kir2.1, Kir7.1 and TASK1 in OL [111,112,113] |
Kir4.1: ↑ Myelination [114][115] | Kir4.1: ↑ Myelination [114,115] | Kir4.1: High level in serum from MS patients [116] |
||||||||||||
| Function | ↑ Maturation [115] | |||||||||||||||||
| Kv | Expression | I | A | and I | K OPC > OL [79][80[118] Kv1.2-6, Kv2.1, Kv7.2 in OPC [110] | OPC > OL [79 | ][81][ | ,80 | 110 | ,81 | 82][117] | ,82 | ] | ,117 | [119][120] Kv4.2, Kv4.3, and Kv3.3. in OPC KCa1.1 in OPC [ | ,118] Kv1.2-6, Kv2.1, Kv7.2 in OPC [110,119,120] Kv4.2, Kv4.3, and Kv3.3. in OPC KCa1.1 in OPC [110] |
Kv mislocalization in EAE animals [121] Kv1.2, Kv1.4, and Kv2.1 downregulated in EAE [122] |
Kv mislocalization in post-mortem human MS lesions [121] |
| Function | ↑ Maturation [87][123] Kv1.3, Kv1.4: ↑ proliferation Kv1.6: ↓ proliferation | ↑ Maturation [87,123] Kv1.3, Kv1.4: ↑ proliferation Kv1.6: ↓ proliferation |
||||||||||||||||
Nav: voltage-gated sodium channels; Cav: voltage-gated calcium channels; Kir: inward-rectifier potassium currents; Kv: voltage-gated potassium channels; IA: transient A-type potassium current; IK: delayed-rectifier potassium currents; KCa: calcium-activated potassium channels; EAE: Experimental Autoimmune Encephalomyelitis; CPZ: cuprizone; MS: Multiple Sclerosis. ↑: increase of. ↓: decrease of.
Nav: voltage-gated sodium channels; Cav: voltage-gated calcium channels; Kir: inward-rectifier potassium currents; Kv: voltage-gated potassium channels; IA: transient A-type potassium current; IK: delayed-rectifier potassium currents; KCa: calcium-activated potassium channels; EAE: Experimental Autoimmune Encephalomyelitis; CPZ: cuprizone; MS: Multiple Sclerosis. ↑: increase of. ↓: decrease of.
Importantlyportantly, some Nav-expressing OPCs can generate APs when stimulated by depolarizing current injection [85][94][124][125][85,94,124,125]. However, OPC-evoked AP has a higher threshold, smaller amplitude and slower kinetics than neuronal counterpart; since synaptic inputs produce only minimal depolarization in NG2+ cells, the physiological relevance of OPC excitability is unclear [110]. However, APs may be involved in a mechanism through which OPCs sense electrically active axons in the environment [94]. In confirm, NG2+ cells receive glutamatergic input upon stimulation from CA3 Schaffer collateral axons in the CA1 region of the hippocampus [28] and, similarly, in the corpus callosum from axons coursing through this region [126][127][126,127]. Synaptic input- and/or Na+ channel-mediated electrical activity may serve as a signal between unmyelinated axonal sections and OPCs that are ready to differentiate into OLs to myelinate these axonal targets [94]. However, not all NG2+ cells with Na+ currents appear to fire regenerative APs, possibly because of low Nav densities [28][95][124][28,95,124]. Hence, Na+ channels are likely to be active upon depolarization in NG2+, probably contributing to the high level of proliferation [95], and could play a role in development and/or CNS repair, e.g., migration. Table 2 summarizes Nav expression and functions in oligodendrogliogenesis.
3.2. Voltage-Gated Ca
2+
Channels in Oligodendroglial Cells
Ca2+ signalling has been shown to regulate many oligodendroglial cell functions, including proliferation, migration, process extension, differentiation, and myelination [86][96][110][136][137][138][139][86,96,110,136,137,138,139]. Intracellular Ca2+ can increase in NG2+ cells throughout several mechanisms, such as direct influx through plasma membrane voltage-gated Ca2+ channels (Cav) or ligand-gated channels [140][141][140,141], as mentioned below, or by the release from internal Ca2+ stores [142][143][142,143] as well as Ca2+-permeable acid-sensing ion channel opening [144].
Cav immunoreactivity was found in CNS white matter, located in oligodendroglial soma, projections and paranodal wraps [145] (see also Table 2). Electrophysiological recordings demonstrated the functional expression of low-voltage and high-voltage activated Ca2+ currents in OPCs from corpus callosum and mouse cortex [81][83][96][97][81,83,96,97], with pharmacological and voltage-dependent properties typical of T-type and L-type Cav, respectively [97]. Indeed, several studies have reported that Ca2+ currents through Cav appear to diminish with maturation of OLs from progenitors to mature cells in culture [83][104][83,104].
These electrophysiological results have been confirmed by RNA-sequencing transcriptome database that revealed high expression of L-type Ca2+ channel subunits in OPCs and subsequent downregulation in newly formed and myelinating OLs [98], indicating that Cav plays a role during the first steps of OPC maturation. The presence of Cav was also confirmed by mRNA analysis for L-type (Cav1.2 and Cav1.3), T-type (Cav3.1 and Cav3.2), P/Q-type (Cav2.1), and N-type (Cav2.2) α1 subunits in NG2+ cells [110]. In particular, Cav1.2 represents the primary pore-forming subunit in OPCs, as siRNA knockdown of Cav1.2 in NG2+ cells removes ∼75% of the Ca2+ elevation following depolarization [96].
Functional Cav are reported to be necessary for glial cell migration in vivo during olfactory glomeruli formation in the developing antennal lobe of sphinx moth Manduca sexta [105]. In addition, in deafferented antennal lobes in which glial cells fail to migrate [106], glial Cav currents are absent, indicating that Cav in glial cells are required to induce or maintain the migration of antennal lobe glial cells into the developing neuropil of the moth [105].
In addition, data indicate that L-type Cav activation, probably mediated by PDGF, contributes to spontaneous Ca2+ oscillations in the OPC soma, leading to accelerated migration and process formation [104]. Furthermore, an increase in amplitude and frequency of Ca2+ transients is one of the mechanisms underlying AMPA-induced stimulation of OPC migration [107], as described below. Furthermore, Ca2+ transients may affect the recycling of cell-adhesion receptors and induce the rearrangement of cytoskeletal components, which are essential for cell movement [108].
When OPCs are grown in high extracellular K+, used as a depolarizing stimulus to activate Cav, they are prompted towards maturation, as demonstrated by a more complex morphology and a significant increase in the expression of mature markers [109]. At the same time, blocking the expression of the Cav α1.2 subunit, that conducts L-type Ca2+ currents, significantly prevents OPC culture maturation [96]. Accordingly, Cav1.2 deficient OPCs present inhibited proliferation and disruption of proliferative response to PDGF, the best known and most active mitogen for OPCs [96]. Interestingly, it was shown that store-operated Ca2+ entry, as well as Ca2+ release from intracellular stores, are essential mechanisms for PDGF-mediated mitotic action in OPCs [86]. A widespread hypothesis is that Ca2+ entry by L-type channels modulates OPC division and cell maturation through independent intracellular pathways. Cav seem to be essential for cell cycle progression of in mitotic OPCs whereas, in post-mitotic pre-OLs, the same channels are playing an important role in cell maturation. In support to this hypothesis, it was demonstrated that a loss of Cav1.2 in oligodendroglial cells affects axonal contact in co-cultured cortical neurons and consequently inhibits the initial steps of myelination [96]. Moreover, deletion of Cav1.2 in OPCs reduces OL maturation and myelination in the postnatal mouse brain and impairs remyelination in a CPZ model [100][101][100,101].
Importantly, it is likely that factors involved in physiological myelination also participate in remyelination of the injured CNS. In this regard, a significant increase in the activity of OPC L-type Cav was found in demyelinated corpus callosum of CPZ-treated mice, suggesting that these channels may play a key role in the induction and/or survival of newly generated OPCs after an insult [102]. Cav expression and functions in oligodendroglial cells are summarized in Table 2.
Cav Channels in Demyelinating Diseases
Several evidences support the notion that aberrant Cav-mediated currents contribute to the pathophysiology of MS or EAE. Increased Ca2+ influx through Cav was assumed to facilitate neurological impairment and histological damage in EAE mice and, by inference, MS [146]. Pregabalin (Lyrica®) is prescribed to MS patients to treat neuropathic pain by targeting Cav [62] and, in addition, it could provide neuroprotection by inhibiting exaggerated Cav currents during excitotoxicity and neuroinflammation. Indeed, it has been demonstrated that Pregabalin treatment alleviates EAE symptoms in mice possibly by reverting, at neuronal level, intracellular Ca2+ overload in EAE lesions [63]. However, in the same paper, the authors pointed to a significant reduction of hippocampal long-term potentiation in pregabalin-treated EAE mice, thus warning of potential side effects on memory and learning processes [58][63][58,63]. In accordance with deleterious effects on memory, two recent clinical studies showed that perioperative pregabalin reduced spatial working memory in humans [64] and its misuse led to cognitive impairment [65].
An abnormal redistribution of N-type Ca2+ channels was found in acutely injured axons, followed by rearrangement of the axonal membrane after injury [147]. Since Nav are known to redistribute along demyelinated axons [46], a similar mechanism may also exist for Cav. Additionally, the N-type Cav2.2 was detected also on mature OLs [99] and the expression of the pore forming α1B-subunit of Cav2.2 was found in MS and EAE plaques and was overexpressed in active lesions [103].
In mice, it has been shown that, after knockout of Cav1.2, axonal myelination is inhibited and OPC maturation disturbed [100]. However, the L-type Ca2+ channel (Cav1.2, Cav1.3, Cav1.4) blockers Bepridil and nitrendipine had comparable beneficial effects in reducing neuroinflammation and axonal pathology on EAE mice [66]. When nimodipine was administered preventively at the time point of disease induction, EAE severity and demyelination decreased [67]. Recently, nimodipine has been reported to have positive effects on Schwann cells, astrocytes and neurons, being associated to increased phosphorylation of either protein kinase B and the cyclic adenosine monophosphate response element-binding protein (CREB) [68][148][68,148]. It is known that axonal Ca2+ overload activates the Ca2+-dependent protease calpain, leading to disruption of the cytoskeleton and to other structural and functional alterations of the axon [149]. Of note, nimodipine also downregulated the expression of calpain as well as the pro-apoptotic protein caspase 3, whereas calbindin expression was upregulated, indicating that modulation of Ca2+ homeostasis and prevention of intracellular Ca2+ overload might be responsible for the neuroprotective properties of this Cav blocker [68][69][68,69].
Interestingly, recent studies have reported that the Cav1.2 channel blockers nimodipine and verapamil exert their neuroprotective effects through anti-inflammatory properties [70], possibly preventing microglial activation [71] and down-regulating TNFα and IL1β expression in the hippocampus [72][73][72,73]. However, microglial cells do not express functional Cav1.2 channels [150], thus the anti-inflammatory effects of these drugs are likely mediated by their block on other cell types [72]. Recently, it was found that animals injected with nimodipine during CPZ-induced demyelination displayed a reduced astrocyte and microglia activation and proliferation as well as a faster and more efficient brain remyelination [74]. Cav1.2 channels are not present in mature OLs [96][104][96,104], but they are expressed by OPCs where they are essential for maturation [100][101][100,101], as mentioned above. This suggests that reducing Cav currents inhibits astrocyte and microglia activation during demyelination. Consequently, pool of proliferating OPC increases as well as the number of myelinating OLs, leading to a beneficial effect for myelin regeneration [74]. However, deletion of the Cav1.2 channels in GFAP+ astrocytes did not prevent myelin damage during CPZ treatment.
Hence, it appears that Cav blockers might represent promising targets for demyelinating diseases as they concur to pathological intracellular Ca2+ overload in neurons and immune cells. Nevertheless, there are no trials for clinical translation of Ca2+ channel blockers so far [151]. For a summary on the effects of Cav-targeting compounds in demyelination see Table 1.
3.3. Voltage-Gated K
+
Channels in Oligodendroglial Cells
The OPC resting membrane potential (Vrest) is near the calculated equilibrium potential for K+ (EK), i.e., −80 mV, suggesting that K+ channels account for the majority of the resting membrane conductance. The predominant K+ channel subtypes open at rest (‘leak’ channels) are the inward-rectifier Kir4.1 and two-pore (K2P) K+ channels [110]. The RNA-Seq transcriptome database shows that Kir4.1 mRNA is expressed at high levels in NG2+ cells [98] (see Table 2). Kir4.1 mediates inward currents observed in NG2+ whole cell recordings upon membrane potential hyperpolarization lower than –100 mV. These currents are blocked by low (200 µM) concentrations of extracellular Ba2+, an inhibitor of Kir channels [110][114][115][110,114,115]. Kir4.1 facilitates clearance of extracellular K+ released during axonal firing, thus maintaining resting membrane potential and AP propagation [46]. Deleting Kir4.1 in mice causes impaired OL maturation and myelination during development, leading to neuronal degeneration [115] and selective deletion of Kir4.1 from OPCs or mature OLs also results in profound functional impairment and axonal degeneration [114][115][114,115].
Outward rectifying voltage-gated K+ channels (Kvs) are also prominent in OPCs, where they are known to regulate cell proliferation and differentiation, and are subsequently downregulated during differentiation [87][123][87,123] (see Table 2). Upon depolarization, NG2+ cells display a non-linear current-to-voltage profile that is shaped by the activation of A-type (IA) and delayed-rectifier (IK) K+ channels. These currents have been extensively characterized in OPCs recorded from cell cultures or in brain slice preparations [79][80][81][82][117][118][79,80,81,82,117,118]. When challenged by a depolarizing voltage step, 4-aminopyridine (4-AP)-sensitive IA contribute to the initial ‘peak’ outward current during the depolarizing phase, due to their rapid activation and inactivation kinetics. Moreover, TEA-sensitive IK, which activate more slowly and do not inactivate, contributes to the sustained ‘steady-state’ current of the depolarizing stimulus [79][80][81][82][117][118][79,80,81,82,117,118]. The relative proportion of the two current components varies by the region of origin. When compared to cortical NG2+ cells, white matter OPCs in P5-P10 mice present higher IK current densities, while IA density is comparable, resulting in a higher IK/IA ratio [124]. During maturation, outward K+ conductances, in particular IK, in OPCs undergo a strong downregulation up to almost completely disappearance in mature OLs [80][87][80,87]. In parallel to IK downregulation, there is a gradual increase in the expression of Kir, that represents the main conductance observed in mature OLs [111], as demonstrated by the Ba2+-sensitivity of overall OPC conductance increases during maturation [152].
RT-PCR and immunocytochemical localization in cultured NG2+ cells have shown robust expression of Kv1.2, Kv1.3, Kv1.4, Kv1.5, Kv1.6 and Kv7.2 mRNA and protein, with Kv1.5 and Kv1.6 showing highest levels among Shaker-type delayed rectifiers [119][120][119,120]. In addition, mRNA for several non-Shaker delayed rectifier channels, Kv7.2 and Kv2.1, are also abundantly expressed [110]. Kv1.4, the only Shaker-type channel to display A-type properties, presents low expression in NG2+ cells database [110]. Other A-type channel subunits that are greatly expressed in OPCs are Kv4.2, Kv4.3, and Kv3.3. Kv1.3 is upregulated during the G1 phase of cell cycle, and blockade of this channel with specific toxins prevents G1/S transition [120]. Conversely, overexpression of Kv1.3 or Kv1.4 promotes OPC proliferation in the absence of mitogens, while overexpression of Kv1.6 inhibits proliferation in the presence of mitogens [123]. On the other hand, neither knockdown nor overexpression of Kv1.5 affect OPC proliferation [119][123][119,123]. Of note, and differently from cell proliferation, differentiation of cultured NG2+ cells into OLs is not significantly affected by overexpression of Kv subunits but is impaired by the Kv blocker TEA [87][109][87,109], demonstrating that oligodendroglial cell proliferation and differentiation might be differently regulated [123].
The large conductance Ca2+-activated (BK) channel KCa1.1 is highly expressed in NG2+ cells [110]. This confirms previous findings that BK channels, which are both voltage- and Ca2+-dependent, are expressed in cultured NG2+ cells [153]. Other K+ channels may also be important in maintaining oligodendroglial cell functions and integrity, including Kir2.1, Kir7.1 and TASK1 channels [111][112][113][111,112,113]. For a summary on Kv and Kir expression in oligodendroglial cells, see Table 2.
Voltage-Gated K+ Channels in Demyelinating Diseases
As mentioned above, deleting Kir4.1 channels in mice causes impaired myelination and OL maturation during development, leading to neuronal degeneration [115]. In confirm, serum levels of antibodies against Kir4.1 were enriched in MS patients, suggesting that Kir4.1 could be a target of antibody responses in this pathology [116].
In myelinated axons, Kv1.1 and Kv1.2 are located underneath the myelin sheath in the paranodes or internodal regions [46], but, after demyelinating insults, alterations in K+ channel expression and distribution along the axon are reported. Indeed, a reduction of Kv1.2, Kv1.4, and Kv2.1 has been demonstrated in EAE mice, correlating with disease severity [122]. Moreover, Calvo et al. found that, although Kv1.1 and Kv1.2 expression levels decrease, they redistributed form the juxtaparanode into the paranode in a spinal nerve transection (SNT) model of neuropathic pain [154]. In contrast, Kv1.4 and 1.6 expression increases within justaparanodes and paranodes [154]. These findings are consistent with previous studies reporting a Kv mislocalization in EAE animals and in post-mortem human MS lesions [121]. In accordance, administration of a Kv1.1 selective blocker (BgK-F6A) ameliorates disease course in EAE mice [75], and treatment with the IA blocker 4-AP enhances axonal conduction [76]. As IA is widely diffused along demyelinated axons and contributes to MS symptoms [155], in 2010 dalfampridine, an extended-release form of 4-AP, has been approved by the FDA to improve walking in MS patients [77]. The mechanism by which Kv blockers could exert neuroprotection is ascribed to the block of excessive K+ outflow from axons causing extracellular K+ homeostasis outbalance that postpones the K+ reversal potential to more positive values. Consequently, a gradual depolarization of neuronal membrane occurs with a consequent inactivation of Nav leading to impaired action potential propagation. Hence, Kv block may help preserving signal conduction. Furthermore, as axonal K+ outflow leads to intracellular K+ depletion causing water loss and disinhibition of proapoptotic enzymes [156], these compounds might also provide neuroprotection by preserving intracellular K+ homeostasis. Of note, a recent paper demonstrated that, although Kv1.4 deficiency decreases OPC proliferation in vitro, it does not influence de- or remyelination in the CPZ model [157]. Moreover, in the same study, Kv1.4 deficiency leads to an ameliorated course of EAE and results in reduced Th1 responses. These data argue for a peripheral effect of Kv1.4 on immune cells, possibly via glial cells. Since the authors did not observe any change in the CPZ model, it is tempting to speculate that the benefits observed in the EAE model are not only due to remyelination, but also to a reduced impact of immune system on the CNS, as discussed previously [158][159][158,159]. For a summary on the effects of Kv-targeting compounds in demyelination see Table 1.