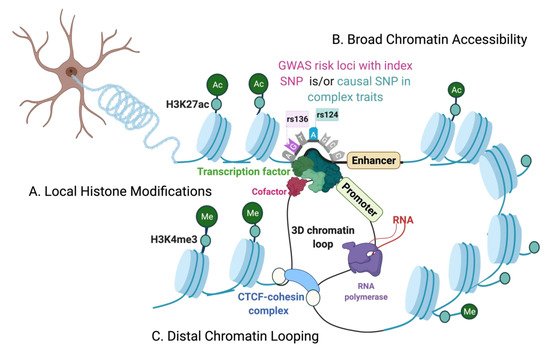
The genetic architecture of complex traits is multifactorial. Genome-wide association studies (GWASs) have identified risk loci for complex traits and diseases that are disproportionately located at the non-coding regions of the genome. On the other hand, we have just begun to understand the regulatory roles of the non-coding genome, making it challenging to precisely interpret the functions of non-coding variants associated with complex diseases. Additionally, the epigenome plays an active role in mediating cellular responses to fluctuations of sensory or environmental stimuli. However, it remains unclear how exactly non-coding elements associate with epigenetic modifications to regulate gene expression changes and mediate phenotypic outcomes. Therefore, finer interrogations of the human epigenomic landscape in associating with non-coding variants are warranted.
- complex traits
- complex diseases
- brain
- non-coding
- epigenome
- DNA structure
- open chromatin
- transcription factors
- histone modifications
- chromatin loops
1. Introduction
The Chromatin Environment
| Epigenomic Features | Techniques | Methods Overview | Benefits | Limitations | Single-Cell and Cell-Types |
|---|---|---|---|---|---|
| 1. Open chromatin regions. 2. Cis-regulatory elements. |
DNase I hypersensitive sites sequencing (DNase-seq). [14] | DNase I digested fragments are extracted using biotin-streptavidin complex. | 1. High signal-to-noise ratio compared to FAIRE-seq. 2. No prior knowledge of locus-specific sequences, primers, or epitope tags is required. 3. Efficiently maps non-coding regions proximal to genes. |
1. DNase I sequence-specific cleavage biases may determine cleavage patterns at the predicted transcription factor (TF) binding sites or footprints. This complicates correctly assessing true transcription factor binding at open chromatin. [15] 2. Requires high number of cells (ideally >= 1 M cells) [14] and a high sequencing depth. 3. Maps relatively low distal regulatory sites compared to formaldehyde-assisted isolation of regulatory elements with sequencing (FAIRE-seq). [16] |
Single-cell (sc)-DNase-seq. [17] |
| 1. Nucleosome positioning. 2. DNA-bound protein binding sites. |
Micrococcal nuclease digestion of chromatin followed by sequencing (MNase-seq) [18], (alternative: nucleosome occupancy and methylome sequencing (NOME-seq). [19] |
Cross-linking to covalently link proteins to the DNA, followed by micrococcal nuclease digestion to remove free DNA. | 1. MNase-seq can map DNA-protein binding for both histone and non-histone proteins. 2. Indirectly maps chromatin accessibility. 3. The digested fraction of accessible chromatin can be repurposed for chromatin immunoprecipitation-based assays (Native-ChIP). |
1. Requires a broad range of sequencing read-out (25 bps to 150 bps) to capture both sub-nucleosome and nucleosome fragments. [20] 2. High dependency on optimized MNase enzyme digestion for reproducibility between experiments. 3. MNase enzyme produces AT cleavage bias that needs bioinformatic corrections. 4. Requires large number of cellular input (ideally >= 1 M cells). |
scMNase-seq, and scNOME-seq. [21,22,23,24] |
| 1. Open chromatin. 2. Cis-regulatory elements. 3. Nucleosome distribution. |
Assay for transposase-accessible chromatin coupled to sequencing (ATAC-seq). [25] | Tn5 transposases-based cutting and tagging of open chromatin. | 1. Low input (ideally <= 50,000 cells) 2. Short and easy to use protocol. 3. Very high signal-to-noise ratio compared to other chromatin accessibility techniques. |
1. Tn5 sequence insertion bias can lead to mapping and/or TF footprinting biases and needs bioinformatic corrections. 2. Mitochondrial contamination of reads (although Omni-ATAC [26] is optimized for lower mitochondrial reads). |
Flow cytometry-based approaches and single cell/nucleus ATAC-seq. [27,28,29,30,31,32] |
| 1. Protein-DNA interactions. 2. Histone post-translational modification. |
Chromatin immunoprecipitation with sequencing (ChIP-seq). [33,34,35] | Formaldehyde crosslinked (X-ChIP) or micrococcal digested fragments (Native-ChIP) followed by immunoprecipitation. | 1. Gold standard to map genome-wide, direct DNA-protein interactions. 2. Single-nucleotide resolution (compared to ChIP-qPCR and ChIP-chip). 3. An ultra-low-input micrococcal nuclease-based native ChIP (ULI-NChIP) can profile genome-wide binding sites of histone proteins with as few as 1000 cells. [36] |
1. Cross-linking and sonication steps (X-ChIP) can lead to high background noise, requiring higher cellular input for optimal signal-to-noise ratio. [33] 2. Relies on the availability and quality of specific antibodies and can suffer from epitope masking due to cross-linking of fragments (X-ChIP). 3. Requires appropriate control experiments to minimize detection of false-positive protein-DNA binding sites. |
sc-ChIP-seq [37] |
| 1. Protein-DNA interactions. 2. Histone post-translational modification. |
ChIP with exonuclease (ChIP-exo) [38], Cleavage under targets & release using nuclease (CUT&RUN) [39], Cleavage under targets and tagmentation. (CUT&TAG) [40] |
ChIP-exo: X-ChIP immunoprecipitated fragments followed by additional λ exonuclease digestion step. CUT&RUN: MNase tethered protein A, targeting specific antibody against the protein of interest. CUT&TAG: Tn5 transposase and protein A fusion protein, targeting antibody against the protein of interest. |
1.ChIP-exo: with an extra exonuclease treatment, it can remove unbound and non-specific DNA, providing higher signal-to-noise ratio over ChIP-seq. [38] 2. CUT&RUN: (i) Uses enzyme-tethering to avoid cross-linking and fragmentation of DNA that greatly reduces the background noise, and epitope masking, making it lower input over ChIP. (ii) It has been validated to map H3K27me3-marked heterochromatin regions. [39] (iii)Use of enzyme-tethering also maps local environment of binding sites, making it suitable to also detect long-range interactions of the protein. 3. CUT&TAG: (i) Requires the least number of cells compared to alternatives (ideally >= 100 cells) and can be performed at single-cell level. [40] (ii) It bypasses cross-linking (compared to ChIP) and library preparation step (compared to ChIP and CUT&RUN). (iii) More sensitive, easier workflow and cost-effective compared to CUT&RUN and alternatives |
1. ChIP-exo: High number of enzymatic steps in ChIP-exo makes it technically challenging and suffers from epitope masking, similar to ChIP. 2.CUT&RUN: (i) Calcium-activated MNase enzyme digestion of chromatin needs to be carefully optimized, to prevent over/under digestion of accessible chromatin. It also relies on antibody quality, like ChIP. (ii) Like X-ChIP, CUT&RUN cannot distinguish direct from indirect 3D contacts. [39] (iii) Requires higher number of cells relative to CUT&TAG (ideally >= 100,000 but can be performed with as low as 1000 cells). [39] 3. CUT&TAG: (i) A potential limitation is antibody-validation, since mapping certain protein-DNA interactions can be more efficient after cross-linking. (ii) Tn5 enzyme biases may confound detection of proteins at heterochromatin regions, since Tn5 preferentially tags accessible chromatin |
CUT&TAG [40] |
| 3. Chromatin loops and 3D interactions. | Chromosome Conformation Capture 3C [41], 4C [42], 5C [43], and Hi-C. [44] |
Formaldehyde cross-linking to covalently link physically interacting chromatin fragments. | 3C/4C/5C: these progressive modifications can map increasingly more chromatin conformations, i.e., one-to-one, one-to-many, and many-to-many epigenetic features, respectively. Hi-C (all-to-all): 1. An unbiased approach that maps genome-wide 3D chromatin conformations. 2. Long-range interactions several mega-base pairs away and high-resolution inter-chromosomal contacts can also be mapped. 3. Low cellular input over 3C/4C (ideally >= 1 M cells). Easy-Hi-C: a biotin-free strategy, more sensitive and requires relatively lower cell input over Hi-C (ideally >= 50,000 cells). [45] |
3C/4C/5C: 1. Maps to a limited resolution and genomic distances of interacting regions. 2. Need priori-defined regions of interests. 3. Cannot resolve long-range contacts by haplotypes (maternal/paternal) of the chromosomes. 4. Requires relatively higher number of cells (ideally >= 10M cells). Hi-C: (i) It cannot detect chromatin contacts with cell-type specificity and cannot detect functional relevance of the chromatin loops. (ii) Some proximity-ligation events can remain undetected due to low efficiency of biotin incorporation at ligation junctions. [45] |
Flow cytometry-based approaches [46,47], sc-Hi-C-seq [48,49], sci-Hi-C-seq [50,51], Dip-C [52] |
| 4. Protein-bound 3D interactions | Chromatin interaction analysis by paired-end tag sequencing (ChIA-PET) [53], HiChIP [54], and Proximity ligation-assisted ChIP-seq (PLAC-seq). [55] |
Formaldehyde cross-linking, followed by antibody-based immunoprecipitation of protein-bound chromatin interactions. | ChIA-PET, HiChIP & PLAC-seq: Can illustrate regulatory roles of 3D chromatin interactions. HiChIP & PLAC-seq: Higher signal-to-noise ratio and significantly lower cell input compared to ChIA-PET. |
ChIA-PET: 1. Low sensitivity in detecting 3D interactions and can have false-positive reads by non-specific antibody binding. 2. Requires very high number of cellular input (ideally >= 100 M cells) [54,56] and high sequencing depth. 3. Ligation of DNA linkers to chromatin fragments can also lead to self-ligation of linkers and false-positive read-outs. ChIA-PET, HiChIP, and PLAC-seq: They all require a priori of target protein of interest and need bioinformatic correction for biases introduced by: ChIP procedure, different fragment lengths, and restriction enzymes cut-site biases. HiChIP and PLAC-seq also require high cell-number (ideally >= 1 M cells). |
Flow cytometry approach [55,57], and multiplex chromatin interaction analysis via droplet-based and barcode-linked sequencing (ChIA-Drop) [58] |
2. Chromatin Accessibility Techniques
2.1. DNase I Hypersensitive Sites Sequencing (DNase-seq)
2.2. Formaldehyde-Assisted Isolation of Regulatory Elements with Sequencing (FAIRE-seq)
2.3. Micrococcal Nuclease Digestion of Chromatin Followed by Sequencing (MNase-seq)
2.4. Assay for Transposase-Accessible Chromatin (ATAC-seq)
References
- Sullivan, P.F.; Daly, M.J.; O’Donovan, M. Genetic architectures of psychiatric disorders: The emerging picture and its implications. Nat. Rev. Genet. 2012, 13, 537–551.
- Tak, Y.G.; Farnham, P.J. Making sense of GWAS: Using epigenomics and genome engineering to understand the functional relevance of SNPs in non-coding regions of the human genome. Epigenet. Chromatin 2015, 8, 57.
- Ripke, S.; Neale, B.M.; Corvin, A.; Walters, J.T.R.; Farh, K.-H.; Holmans, P.A.; Lee, P.; St Clair, D.; Weinberger, D.R.; Wendland, J.R.; et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427.
- O’Connor, L.J.; Schoech, A.P.; Hormozdiari, F.; Gazal, S.; Patterson, N.; Price, A.L. Extreme polygenicity of complex traits is explained by negative selection. Am. J. Hum. Genet. 2019, 105, 456–476.
- The ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74.
- Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Milosavljevic, A.; Ren, B.; Stamatoyannopoulos, J.A.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330.
- Cavalli, G.; Heard, E. Advances in epigenetics link genetics to the environment and disease. Nature 2019, 571, 489–499.
- Lutz, P.-E.; Tanti, A.; Gasecka, A.; Barnett-Burns, S.; Kim, J.J.; Zhou, Y.; Chen, G.G.; Wakid, M.; Shaw, M.; Almeida, D.; et al. Association of a history of child abuse with impaired myelination in the anterior cingulate cortex: Convergent epigenetic, transcriptional, and morphological evidence. AJP 2017, 174, 1185–1194.
- Labonté, B.; Suderman, M.; Maussion, G.; Navaro, L.; Yerko, V.; Mahar, I.; Bureau, A.; Mechawar, N.; Szyf, M.; Meaney, M.J.; et al. Genome-wide epigenetic regulation by early-life trauma. Arch. Gen. Psychiatry 2012, 69, 722–731.
- Patrick, E.; Taga, M.; Ergun, A.; Ng, B.; Casazza, W.; Cimpean, M.; Yung, C.; Schneider, J.A.; Bennett, D.A.; Gaiteri, C.; et al. Deconvolving the contributions of cell-type heterogeneity on cortical gene expression. PLoS Comput. Biol. 2020, 16, e1008120.
- Li, G.; Levitus, M.; Bustamante, C.; Widom, J. Rapid spontaneous accessibility of nucleosomal DNA. Nat. Struct. Mol. Biol. 2005, 12, 46–53.
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395.
- DDD Study; Konrad, E.D.H.; Nardini, N.; Caliebe, A.; Nagel, I.; Young, D.; Horvath, G.; Santoro, S.L.; Shuss, C.; Ziegler, A.; et al. CTCF variants in 39 individuals with a variable neurodevelopmental disorder broaden the mutational and clinical spectrum. Genet. Med. 2019, 21, 2723–2733.
- Gallegos, D.A.; Chan, U.; Chen, L.-F.; West, A.E. chromatin regulation of neuronal maturation and plasticity. Trends Neurosci. 2018, 41, 311–324.
- Pang, B.; Qiao, X.; Janssen, L.; Velds, A.; Groothuis, T.; Kerkhoven, R.; Nieuwland, M.; Ovaa, H.; Rottenberg, S.; van Tellingen, O.; et al. Drug-induced histone eviction from open chromatin contributes to the chemotherapeutic effects of doxorubicin. Nat. Commun. 2013, 4, 1908.
- Crawford, G.E.; Davis, S.; Scacheri, P.C.; Renaud, G.; Halawi, M.J.; Erdos, M.R.; Green, R.; Meltzer, P.S.; Wolfsberg, T.G.; Collins, F.S. DNase-Chip: A high-resolution method to identify DNase I hypersensitive sites using tiled microarrays. Nat. Methods 2006, 3, 503–509.
- Song, L.; Crawford, G.E. DNase-Seq: A high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb. Protoc. 2010, 2010.
- Dorschner, M.O.; Hawrylycz, M.; Humbert, R.; Wallace, J.C.; Shafer, A.; Kawamoto, J.; Mack, J.; Hall, R.; Goldy, J.; Sabo, P.J.; et al. High-throughput localization of functional elements by quantitative chromatin profiling. Nat. Methods 2004, 1, 219–225.
- Maurano, M.T.; Humbert, R.; Rynes, E.; Thurman, R.E.; Haugen, E.; Wang, H.; Reynolds, A.P.; Sandstrom, R.; Qu, H.; Brody, J.; et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 2012, 337, 1190–1195.
- Funk, C.C.; Casella, A.M.; Jung, S.; Richards, M.A.; Rodriguez, A.; Shannon, P.; Donovan-Maiye, R.; Heavner, B.; Chard, K.; Xiao, Y.; et al. Atlas of transcription factor binding sites from ENCODE DNase hypersensitivity data across 27 tissue types. Cell Rep. 2020, 32, 108029.
- Sung, M.-H.; Guertin, M.J.; Baek, S.; Hager, G.L. DNase footprint signatures are dictated by factor dynamics and DNA sequence. Mol. Cell 2014, 56, 275–285.
- Shibata, Y.; Sheffield, N.C.; Fedrigo, O.; Babbitt, C.C.; Wortham, M.; Tewari, A.K.; London, D.; Song, L.; Lee, B.-K.; Iyer, V.R.; et al. Extensive evolutionary changes in regulatory element activity during human origins are associated with altered gene expression and positive selection. PLoS Genet. 2012, 8, e1002789.
- Lu, Y.; Wang, X.; Yu, H.; Li, J.; Jiang, Z.; Chen, B.; Lu, Y.; Wang, W.; Han, C.; Ouyang, Y.; et al. Evolution and comprehensive analysis of DNaseI hypersensitive sites in regulatory regions of primate brain-related genes. Front. Genet. 2019, 10, 152.
- ReproGen Consortium; Schizophrenia Working Group of the Psychiatric Genomics Consortium; The RACI Consortium; Finucane, H.K.; Bulik-Sullivan, B.; Gusev, A.; Trynka, G.; Reshef, Y.; Loh, P.-R.; Anttila, V.; et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 2015, 47, 1228–1235.
- Jin, W.; Tang, Q.; Wan, M.; Cui, K.; Zhang, Y.; Ren, G.; Ni, B.; Sklar, J.; Przytycka, T.M.; Childs, R.; et al. Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature 2015, 528, 142–146.
- Giresi, P.G.; Kim, J.; McDaniell, R.M.; Iyer, V.R.; Lieb, J.D. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 2007, 17, 877–885.
- Song, L.; Zhang, Z.; Grasfeder, L.L.; Boyle, A.P.; Giresi, P.G.; Lee, B.-K.; Sheffield, N.C.; Graf, S.; Huss, M.; Keefe, D.; et al. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res. 2011, 21, 1757–1767.
- Koues, O.I.; Kowalewski, R.A.; Chang, L.-W.; Pyfrom, S.C.; Schmidt, J.A.; Luo, H.; Sandoval, L.E.; Hughes, T.B.; Bednarski, J.J.; Cashen, A.F.; et al. Enhancer sequence variants and transcription-factor deregulation synergize to construct pathogenic regulatory circuits in B-cell lymphoma. Immunity 2015, 42, 186–198.
- Davie, K.; Jacobs, J.; Atkins, M.; Potier, D.; Christiaens, V.; Halder, G.; Aerts, S. Discovery of transcription factors and regulatory regions driving in vivo tumor development by ATAC-Seq and FAIRE-Seq open chromatin profiling. PLoS Genet. 2015, 11, e1004994.
- Simon, J.M.; Giresi, P.G.; Davis, I.J.; Lieb, J.D. Using FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) to isolate active regulatory DNA. Nat. Protoc. 2012, 7, 256–267.
- Kelly, T.K.; Liu, Y.; Lay, F.D.; Liang, G.; Berman, B.P.; Jones, P.A. Genome-wide mapping of nucleosome positioning and DNA methylation within individual DNA molecules. Genome Res. 2012, 22, 2497–2506.
- Voong, L.N.; Xi, L.; Wang, J.-P.; Wang, X. Genome-wide mapping of the nucleosome landscape by micrococcal nuclease and chemical mapping. Trends Genet. 2017, 33, 495–507.
- Ozsolak, F.; Song, J.S.; Liu, X.S.; Fisher, D.E. High-throughput mapping of the chromatin structure of human promoters. Nat. Biotechnol. 2007, 25, 244–248.
- Ramachandran, S.; Ahmad, K.; Henikoff, S. Transcription and remodeling produce asymmetrically unwrapped nucleosomal intermediates. Mol. Cell 2017, 68, 1038–1053.e4.
- Henikoff, J.G.; Belsky, J.A.; Krassovsky, K.; MacAlpine, D.M.; Henikoff, S. Epigenome characterization at single base-pair resolution. Proc. Natl. Acad. Sci. USA 2011, 108, 18318–18323.
- Yazdi, P.G.; Pedersen, B.A.; Taylor, J.F.; Khattab, O.S.; Chen, Y.-H.; Chen, Y.; Jacobsen, S.E.; Wang, P.H. Nucleosome organization in human embryonic stem cells. PLoS ONE 2015, 10, e0136314.
- Teif, V.B.; Vainshtein, Y.; Caudron-Herger, M.; Mallm, J.-P.; Marth, C.; Höfer, T.; Rippe, K. Genome-wide nucleosome positioning during embryonic stem cell development. Nat. Struct. Mol. Biol. 2012, 19, 1185–1192.
- Kundaje, A.; Kyriazopoulou-Panagiotopoulou, S.; Libbrecht, M.; Smith, C.L.; Raha, D.; Winters, E.E.; Johnson, S.M.; Snyder, M.; Batzoglou, S.; Sidow, A. Ubiquitous heterogeneity and asymmetry of the chromatin environment at regulatory elements. Genome Res. 2012, 22, 1735–1747.
- Clark, S.C.; Chereji, R.V.; Lee, P.R.; Fields, R.D.; Clark, D.J. Differential nucleosome spacing in neurons and glia. Neurosci. Lett. 2020, 714, 134559.
- Berkowitz, E.M.; Sanborn, A.C.; Vaughan, D.W. Chromatin structure in neuronal and neuroglial cell nuclei as a function of age. J. Neurochem. 1983, 41, 516–523.
- Druliner, B.R.; Vera, D.; Johnson, R.; Ruan, X.; Apone, L.M.; Dimalanta, E.T.; Stewart, F.J.; Boardman, L.; Dennis, J.H. Comprehensive nucleosome mapping of the human genome in cancer progression. Oncotarget 2015, 7, 13429–13445.
- Chutake, Y.K.; Costello, W.N.; Lam, C.; Bidichandani, S.I. Altered nucleosome positioning at the transcription start site and deficient transcriptional initiation in friedreich ataxia. J. Biol. Chem. 2014, 289, 15194–15202.
- Sun, H.; Damez-Werno, D.M.; Scobie, K.N.; Shao, N.-Y.; Dias, C.; Rabkin, J.; Koo, J.W.; Korb, E.; Bagot, R.C.; Ahn, F.H.; et al. ACF chromatin-remodeling complex mediates stress-induced depressive-like behavior. Nat. Med. 2015, 21, 1146–1153.
- Goodman, J.V.; Bonni, A. Regulation of neuronal connectivity in the mammalian brain by chromatin remodeling. Curr. Opin. Neurobiol. 2019, 59, 59–68.
- Ronan, J.L.; Wu, W.; Crabtree, G.R. From neural development to cognition: Unexpected roles for chromatin. Nat. Rev. Genet. 2013, 14, 347–359.
- Li, C.; Luscombe, N.M. Nucleosome positioning stability is a significant modulator of germline mutation rate variation across the human genome. Genomics 2018. preprint.
- Gao, W.; Lai, B.; Ni, B.; Zhao, K. Genome-wide profiling of nucleosome position and chromatin accessibility in single cells using scmnase-seq. Nat. Protoc. 2020, 15, 68–85.
- Lai, B.; Gao, W.; Cui, K.; Xie, W.; Tang, Q.; Jin, W.; Hu, G.; Ni, B.; Zhao, K. Principles of nucleosome organization revealed by single-cell micrococcal nuclease sequencing. Nature 2018, 562, 281–285.
- Pott, S. Simultaneous measurement of chromatin accessibility, DNA methylation, and nucleosome phasing in single cells. eLife 2017, 6, e23203.
- Clark, S.J.; Argelaguet, R.; Kapourani, C.-A.; Stubbs, T.M.; Lee, H.J.; Alda-Catalinas, C.; Krueger, F.; Sanguinetti, G.; Kelsey, G.; Marioni, J.C.; et al. ScNMT-Seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nat. Commun. 2018, 9, 781.
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218.
- Corces, M.R.; Trevino, A.E.; Hamilton, E.G.; Greenside, P.G.; Sinnott-Armstrong, N.A.; Vesuna, S.; Satpathy, A.T.; Rubin, A.J.; Montine, K.S.; Wu, B.; et al. An improved ATAC-Seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 2017, 14, 959–962.
- Bryois, J.; Garrett, M.E.; Song, L.; Safi, A.; Giusti-Rodriguez, P.; Johnson, G.D.; Shieh, A.W.; Buil, A.; Fullard, J.F.; Roussos, P.; et al. Evaluation of chromatin accessibility in prefrontal cortex of individuals with schizophrenia. Nat. Commun. 2018, 9, 3121.
- Fullard, J.F.; Hauberg, M.E.; Bendl, J.; Egervari, G.; Cirnaru, M.-D.; Reach, S.M.; Motl, J.; Ehrlich, M.E.; Hurd, Y.L.; Roussos, P. An atlas of chromatin accessibility in the adult human brain. Genome Res. 2018, 28, 1243–1252.
- Trevino, A.E.; Sinnott-Armstrong, N.; Andersen, J.; Yoon, S.-J.; Huber, N.; Pritchard, J.K.; Chang, H.Y.; Greenleaf, W.J.; Pașca, S.P. Chromatin accessibility dynamics in a model of human forebrain development. Science 2020, 367, eaay1645.
- Hauberg, M.E.; Creus-Muncunill, J.; Bendl, J.; Kozlenkov, A.; Zeng, B.; Corwin, C.; Chowdhury, S.; Kranz, H.; Hurd, Y.L.; Wegner, M.; et al. Common schizophrenia risk variants are enriched in open chromatin regions of human glutamatergic neurons. Nat. Commun. 2020, 11, 5581.
- Buenrostro, J.D.; Wu, B.; Litzenburger, U.M.; Ruff, D.; Gonzales, M.L.; Snyder, M.P.; Chang, H.Y.; Greenleaf, W.J. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 2015, 523, 486–490.
- Ziffra, R.S.; Kim, C.N.; Wilfert, A.; Turner, T.N.; Haeussler, M.; Casella, A.M.; Przytycki, P.F.; Kreimer, A.; Pollard, K.S.; Ament, S.A.; et al. Single cell epigenomic atlas of the developing human brain and organoids. Dev. Biol. 2019. preprint.
- Cusanovich, D.A.; Daza, R.; Adey, A.; Pliner, H.A.; Christiansen, L.; Gunderson, K.L.; Steemers, F.J.; Trapnell, C.; Shendure, J. Multiplex single-cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 2015, 348, 910–914.
- Li, Y.E.; Preissl, S.; Hou, X.; Zhang, Z.; Zhang, K.; Fang, R.; Qiu, Y.; Poirion, O.; Li, B.; Liu, H.; et al. An atlas of gene regulatory elements in adult mouse cerebrum. Neuroscience 2020. preprint.
- Corces, M.R.; Shcherbina, A.; Kundu, S.; Gloudemans, M.J.; Frésard, L.; Granja, J.M.; Louie, B.H.; Eulalio, T.; Shams, S.; Bagdatli, S.T.; et al. Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson’s diseases. Nat. Genet. 2020, 52, 1158–1168.
- Mulqueen, R.M.; DeRosa, B.A.; Thornton, C.A.; Sayar, Z.; Torkenczy, K.A.; Fields, A.J.; Wright, K.M.; Nan, X.; Ramji, R.; Steemers, F.J.; et al. Improved single-cell ATAC-Seq reveals chromatin dynamics of in vitro corticogenesis. Genomics 2019. preprint.
- Lake, B.B.; Chen, S.; Sos, B.C.; Fan, J.; Kaeser, G.E.; Yung, Y.C.; Duong, T.E.; Gao, D.; Chun, J.; Kharchenko, P.V.; et al. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat. Biotechnol. 2018, 36, 70–80.
- Chen, S.; Lake, B.B.; Zhang, K. High-throughput sequencing of the transcriptome and chromatin accessibility in the same cell. Nat. Biotechnol. 2019, 37, 1452–1457.