Cannabinoquinones are highly electrophilic compounds and undergo non-transient thia-Michael addition when assayed in the cysteamine assay (). With 2-substituted compounds, the assay was complicated by the interaction of the amino group of cysteamine with the quinone carbonyl, resulting in a change of the DMSO spectrum upon cysteamine addition, presumably due to the transient Schiff base formation.
- cannabinoids
- quinones
- oxidation
- thia-Michael addition
- immunomodulation
- scleroderma
1. Introduction
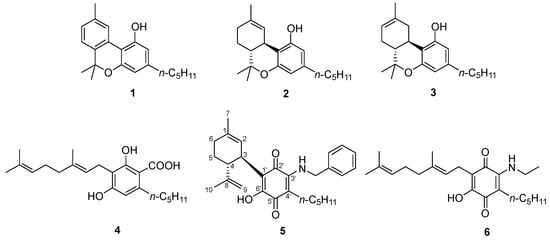
Phytocannabinoids are endogenously produced by Cannabis sativa L. in an acidic form, a discovery reported by Krejčí and Šantavý [1] five decades after the seminal purification of cannabinol (CBN, Figure 1 , 1) from hashish in 1899 [2]. Exogenous factors (heat, light, oxygen) are next responsible for the non-enzymatic expansion of the diversity of the native phytocannabinoid pool [3]. Neutral cannabinoids are generated by decarboxylation of their acidic precursors, a reaction dependent on the presence of a mesomerically electron-donating para -oxygen atom [4], while atmospheric oxidation can affect both the terpenyl residue, as shown by the aromatization of Δ 9- and Δ 8-tetrahydrocannabinols [Δ 9- and Δ 8- THC ( 2, 3, respectively) to cannabinol ( 1) [3], and the resorcinyl core, generating colored quinones [5]. Air oxidation of resorcinyl phytocannabinoids is relatively slow, at least in the plant material and in the solid state, and cannabinoquinones are susceptible to further oxidation and degradation. Compounds of this type have, therefore, only been isolated as trace constituents of C. sativa extracts [3]. Nevertheless, they have played an important role in the history of Cannabis , since their formation provided the basis for the Beam test, the first forensic color assay for hashish [6], and giving a reddish hue to the high vacuum Cannabis distillates ( red oils ) from which the first phytocannabinoids were isolated and characterized [7]. The lack of narcotic activity of acidic cannabinoids, biogenetically derived from cannabigerolic acid (CBGA, 4), and of cannabinoquinones, has long been reductively equated to a lack of bioactivity. This and instability issues have combined to delay systematic studies in the area, but significant clinical potential has now emerged for both classes of compounds, and especially cannabinoquinones. Thus, the aminocannabinoquinone VCE-004.8 ( 5) is currently in phase 2 clinical studies with orphan drug status in the EU and USA for the treatment of diffuse cutaneous systemic sclerosis [8]; its analogue VCE-003.2 ( 6) has completed pre-clinical investigation [9], receiving orphan drug designation by the EMA and FDA for the management of Huntington’s disease, and interest in the anticancer activity of cannabinoquinones has been revamped by recent studies [10].
2. Synthesis and Chemistry of Cannabinoquinoids
The formation of cannabinoquinones under non-radical conditions was first reported by Mechoulam, who discovered that the λ 3-periodinane bis(trifluoroacetoxy)iodobenzene (BTIP, PIFA) could convert Δ 8-THC (3) into the quinone 12 [11][16]. The reaction involves formation of an iodonium (III) intermediate, that next undergoes nucleophilic attack by an external water molecule to the para -position, less encumbered compared to the ortho -positions. Elimination of iodobenzene eventually generates a p -quinone in yields between 31% and 50% ( Scheme 3 ).
Unexpectedly, oxidation of the monophenolic phytocannabinoid cannabichromene (CBC, 42) afforded a p -quinone ( 44) also with SIBX [12][21], presumably because of electrocyclic opening of the chromene ring of an originally generated o -quinone ( 43) to a cyclohexentrione ( 44), that by a series of keto-enolic tautomerization steps undergoes electrocyclizative aromatization to a p -quinone ( Scheme 5 ) [13][22].
Acidic cannabinoids are stable under Beam-conditions, and did not afford quinones upon reaction with SIBX, that, just like the Beam biphasic system [9], could oxidize 2-alkyl substituted cannabinoids to their corresponding quinones. Taken together, the results of these synthetic studies have established iodanes as the oxidants of choice for the generation of cannabinoquinones from phytocannabinoids. The λ 5-iodane SIBX can be used for diphenolic substrates, while the λ 3-iodane PIFA is required for monophenolic phytocannabinoids, with the notable exception of chromenic substrates, where valence equilibration converts o -quinones into p -quinones [12][21]. The dimerization that plagues the autoxodidative reactions is not observed with iodane oxidants, and any potential issue of metal retention by chelation is also prevented. The iodinane oxidation of resorcinolic cannabinoids could be scaled-up to gram-levels [5], and proved suitable for the GMP-production of the amounts of VCE-004.8 required for the phase 1 clinical studies [14][23].
Cannabinoquinones are highly electrophilic compounds and undergo non-transient thia-Michael addition when assayed in the cysteamine assay ( Scheme 7 ) [15][24]. With 2-substituted compounds, the assay was complicated by the interaction of the amino group of cysteamine with the quinone carbonyl, resulting in a change of the DMSO spectrum upon cysteamine addition, presumably due to the transient Schiff base formation. This resulted in chemical shift changes, with no evidence, however, of thiol addition [15][24]. Phase switch to CDCl 3 or extraction with hexane regenerated the starting compound, showing that the introduction of a substituent at the 2-position quenches Michael reactivity, an observation of relevance for the bioactivity profile of cannabinoquinones (see Section 5.1 ).
3. Spectroscopic Properties of Cannabinoquinones
Parallel to the color development, NMR spectroscopy can be an efficient system to detect and monitor the conversion of phytocannabinoids into the corresponding monomeric quinones. 1H NMR spectra of p -cannabinoquinones are characterized by the obvious loss of one of the two resorcinyl protons, with the remaining signal experiencing a downfield shift of ab. 0.2–0.3 ppm ( Figure 27 ) [5]. The hallmark of p -cannabinoquinones 13C NMR spectra is the presence of the two carbonyl resonances around δ C 180–190 ppm. The third oxygen-bearing carbon of the same ring resonates at ~δ C 150 ppm, while the single methine experiences a marked downfield shift to ~δ C 130 ppm [5][12][5,21].
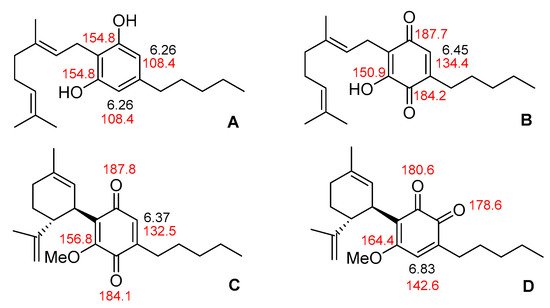
As described in Section 3.2.2 , a different order in the methylation/oxidation sequence can provide alternative ortho - and para - O -methyl cannabinoquinones, providing the opportunity of a vis-à-vis spectroscopic comparison between isomeric quinoids. As shown in Figure 27 , the single proton signal of the quinone ring is markedly downfield shifted ( Δδ ab. 0.4 ppm) in o -cannabinoquinones and other diagnostic differences are also present in the 13C NMR spectra. Indeed, the ortho - isomer can be easily identified, since the couple of carbonyl signals resonates at significantly lower ppm values compared to the para - one (δ C 175–180 vs. δ C 185–190) [13][22]. Both this high-field shift and the higher resonance values of the -OMe-bearing carbon can be explained by considering that an ortho -quinone carbonyl is a better electron sink than a para -quinone carbonyl for the mesomeric delocalization of the lone pair of the methoxy oxygen. Thus, due to a more extended conjugation, the contribution of the zwitterionic resonance form that includes a methoxonium ion is larger in ortho -cannabinoquinones compared to para -cannabinoquinones ( Figure 8 ).
4. Bioactivity Studies
Taken together, these studies show that cannabinoquinones are interesting structures for the development of novel anticancer agents, but the stability in solution of the lead compound of the class (CBDQ, HU-331, 9) seems too limited for further developments. In addition, the mechanism of activity needs additional investigation, while the pharmacokinetic profile of these compounds is still unknown.
Interest in cannabinoquinones in the neuro-immunological area was spurred by considerations of the biological profile of CBD ( 7). This compound has a multitude of low-affinity targets that includes GPCR, ion channels, enzymes, and transcription factors of critical relevance for neuroprotection and immunity [16][35]. The twisted bicyclic structural element of CBD seems to have an inherent, broadly tuned complementarity for biological surfaces, possibly expressed by different torsion angles along the methyl-aryl atropoisomeric axis, unable, however, to engage the compound in tight intermolecular interactions. In this context, the increased polarization associated to the resorcinol-to-hydroxyquinone oxidation is expected to strengthen electrostatic and hydrogen bonding interactions with macromolecular targets, additionally increasing biological “stickiness” by metal chelation, covalent reactivity via Michael addition and involvement in redox cycles. This strategy was confirmed by comparison of the PPAR-γ modulation activity of cannabidiol (CBD) and its corresponding quinone (CBDQ). While the former showed only marginal activity in the assay (EC 50 > 25 μM), the latter was endowed with potent activity (EC 50 = 10.5 μM), further increased by one order of magnitude by the aza-Michael adducts [8]. These observations were duplicated with the quinones of cannabigerol (CBG, 13) and CBC ( 42) [9], suggesting a class-specific trend, that led to extensive studies and to the selection of two candidates for systematic studies.
The CBG-derived amino-hydroxyquinone 6 (VCE-003.2) showed potent agonistic activity at PPAR-γ (IC 50 = 1.2 μM), binding both the canonical and the alternative binding sites of the transcription factor, and lacking activity on cannabinoid receptors [17][36]. In BV2 mice microglia- and astrocytes cells, 6 could inhibit the secretion of various soluble pro-inflammatory mediators (IL-6, TNF-α, IL-1b) at one-digit μM concentration, reducing the expression of iNOS in BV2 microglia cells in a way that is sensitive to PPAR-γ (GW9662) antagonists [17][36]. When orally or intraperitoneally administered, 6 showed beneficial effects in murine models of Huntington’s disease [9][18][9,37], Parkinson’s disease [17][19][36,38], and amyotrophic lateral sclerosis [20][39]. Compound 6, formulated as EHP-102, has completed preclinical development and is entering a phase 1 study for Huntington’s disease and Parkinson’s disease, with orphan drug status in the EU and USA for the former condition [21][40].
Within the aminoquinones derived from CBD ( 7), the benzylamine adduct 5 emerged as a powerful activator of the PPAR-γ transcriptional activity [8] and, of the hypoxia inducible factor (HIF-1) pathway, an activity resulting from inhibition of prolyl hydroxylases [22][41]. This aminoquinone 5 (VCE-004.8) was neutral at CB 1 but behaved as an activator at CB 2 [8], and was a partial PPAR-γ activator, devoid of adipogenic activity. In a murine model of metabolic syndrome (high-fat diet), 5 could reduce the metabolic disruption and inflammation associated to this condition, promoting weight loss, and reducing total fat mass, adipocyte volume, and plasma triglycerides [23][42]. Remarkably, it could also increase FGF21 mRNA expression in white and brown adipose cells, as well as in a BAT cell, significantly ameliorating glucose tolerance, reducing leptin levels and increasing adiponectin and incretins ( GLP-1, GIP) levels [23][42]. VCE-004.8 ( 5) showed anti-fibrotic activity in a murine scleroderma model as well as in different multiple sclerosis models, preventing immune cell infiltration, demyelination, and axonal damage [24][43]. Its oral formulation (EHP-101) has completed a Phase 1 study on over 100 healthy volunteers, demonstrating excellent safety and tolerability, and is proceeding now, under orphan drug status by EMA in Europe and FDA in USA, to a Phase 2 clinical study for the management of scleroderma [25][44].