Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Fabio Del Bello and Version 2 by Vivi Li.
Levodopa (LD) is the most effective drug in the treatment of Parkinson’s disease (PD). However, although it represents the “gold standard” of PD therapy, LD can cause side effects, including gastrointestinal and cardiovascular symptoms as well as transient elevated liver enzyme levels. Moreover, LD therapy leads to LD-induced dyskinesia (LID), a disabling motor complication that represents a major challenge for the clinical neurologist. Due to the many limitations associated with LD therapeutic use, other dopaminergic and non-dopaminergic receptor drugs, including serotoninergic, gluamatergic and noradrenergic receptor ligands, are being developed to optimize the treatment response.
- Parkinson’s disease
- levodopa therapy
- levodopa-induced side effects
- dopaminergic drugs
- non-dopaminergic receptor ligands
1. Introduction
Parkinson’s disease (PD), also known as idiopathic paralysis agitans, is one of the most frequent chronic neurodegenerative diseases worldwide. Although its etiology has not been determined so far, the main pathological characteristic is the decrease of the dopamine (DA) level due to the degeneration of the dopaminergic neurons in the substantia nigra pars compacta [1][2][1,2]. This leads to motor (i.e., postural instability, dyskinesias, tremor, and rigidity) and non-motor (i.e., depression, cognitive impairment, pain, hallucinations) symptoms [3][4][5][6][7][8][9][10][11][12][13][3,4,5,6,7,8,9,10,11,12,13]. Another pathologically severe aspect is the abnormal formation of protein aggregates inside nerve cells (Lewy bodies), whose primary structural component is the presynaptic neuronal protein α-synuclein. For this reason, PD is classified as synucleopathy. Unfortunately, effective inhibition of progression or the cure for PD is not yet available, while all the available therapies only provide relief for symptoms.
Dopaminergic medications are currently the most effective treatment for both motor and non-motor symptoms, though they are not devoid of limitations and frequently produce undesired side effects. The standard treatment of PD patients consists in the administration of DA) in the form of levodopa (LD), a catecholamine produced by the intraneuronal tyrosine hydroxylation [14][15][16][17][18][19][14,15,16,17,18,19]. Its combination with a peripheral DOPA decarboxylase inhibitor (i.e., carbidopa) increases LD availability in the central nervous system (CNS) and ameliorates the therapeutic profile of LD, prolonging its efficacy [20][21][22][20,21,22]. An increase in the efficacy of dopaminergic therapy is also obtained by the simultaneous blockade of the DA metabolism with monoaminooxidase B (MAO-B) and/or catechol-O-methyl transferase (COMT) inhibitors [23][24][23,24]. Although LD represents the “gold standard” of PD therapy [25], unfortunately, orally administered LD can cause side effects, including gastrointestinal and cardiovascular symptoms as well as transient elevated liver enzyme levels. Moreover, LD therapy leads to LD-induced dyskinesia (LID) [26], a disabling motor complication that represents a major challenge for the clinical neurologist [27]. Indeed, LID negatively affects the quality of life [28][29][30][28,29,30] and constitutes a serious obstacle to the management of PD imposing a limit and a reduction of LD dosage, thus restricting treatment efficacy [27].
Numerous therapies are currently being developed to treat the motor and non-motor complications of PD and LID [31].
Mostly a customized combination of DA agonists and LD formulations is performed. The striatal D1 and D2 receptors are the common binding sites of DA ligands for PD treatment, but lately D3 and D4 subtypes have also become potential targets.
At the best of our knowledge, ten DA agonists are so far available for this disease. They can be listed in ergot DA agonists, including Bromocriptine, Cabergoline, Dihydroergocriptine, Lisuride, and Pergolide and non-ergot DA agonists, including Piribedil, Pramipexole, Ropinirole, Apomorphine, and Rotigotine [32] (Figure 1).
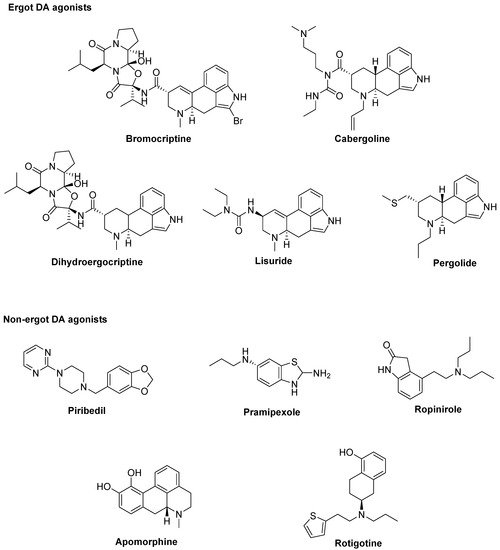
Figure 1. Dopamine (DA) agonists available for Parkinson’s disease (PD) treatment.
Unluckily, DA agonists are not devoid of significant side effects such as hallucinations, hypotension, nausea, vomiting, pathological gambling, compulsive shopping and hypersexuality [32][33][33,34]. As a therapeutic example, symptoms of early stage PD may be controlled by the treatment with Pramipexole [34][35], but after a while a combination with LD is needed to optimize the management of PD symptoms [35][36]. Thus, DA agonists are typically used either to reduce the dosage of LD or to delay its use (LD sparing) [36][37], although it has been discussed that dyskinesia evolvement is due to disease persistence rather than protracted LD use [37][38]. Recently, research on dopaminergic targets has produced some new interesting candidates (Figure 2).
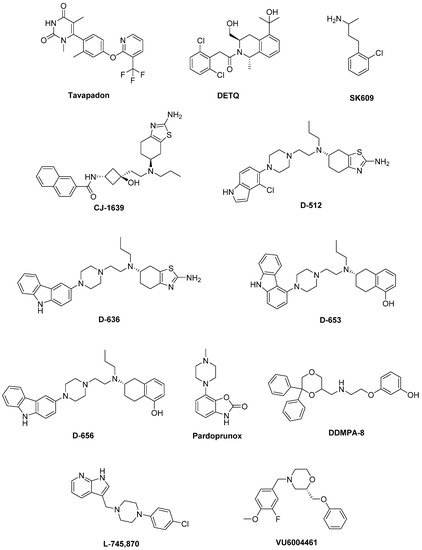
Figure 2. Emerging dopaminergic ligands as new levodopa (LD) adjuvant candidates.
Among these, Tavapadon (or PF-06649751) is a novel, highly selective D1/D5 agonist. A recent paper, reporting about Phase I PD studies, candidates Tavapadon as a novel therapeutic agent for PD with an initial safety, tolerability, and pharmacokinetic profile as well as potential for efficacy. The same report asserts that Phase II clinical trials have been initiated to deeper investigate the potential safety and efficacy of Tavapadon with the aim to determine the dose that can produce relief of symptoms while reducing dependence on LD and, in the meanwhile, avoiding the problems associated with long-term LD administration [38][39].
Preclinical and clinical studies have indicated the potential utility of D1 agonists for the treatment of neuropsychiatric disorders. However, these agents are not devoid of limitations. For instance, it has been demonstrated that LID results from increased D1 receptor-mediated transmission at the level of the direct pathway. Moreover, unlike positive allosteric modulators (PAMs), orthosteric D1 receptor agonists produce receptor desensitization and an inverted U-shaped dose-response curve [39][40]. The development of the D1 PAM DETQ has been reported as a different approach to D1 receptor activation [40][41]. Being able to amplify the effects of released endogenous DA in situ, DETQ gives a more physiological response. Its CNS pharmacology strictly reminds that of D1 agonists, but also shows remarkable differences (i.e., it does not induce stereotypy or desensitization) [40][41]. The reported behavioral and neurochemical test results suggest a therapeutic utility in neuropsychiatric disorders such as PD [41][42][42,43].
It has also been hypothesized that DA receptors in the striatum can form heteromeric complexes. Such an heteromerization leads to changes in the functional and pharmacological properties of receptors compared to their monomeric subtypes [43][44][44,45]. It has been observed a correlation between the expression of D1–D3 receptor heteromers and the development of LID [45][46]. Furthermore, D3 receptor stimulation can potentiate the D1 receptor signaling pathway [45][46][46,47]. Thus, future D3 antagonists or partial agonists able to selectively modulate the activity of striatal D1–D3 receptor heteromers could be very promising in LID control [47][48]. Treatment with LD also induces an ectopic expression of D3 receptors in the DA depleted dorsal striatum, which is associated with dyskinesia [39][45][48][40,46,49]. D3 receptor involvement in dyskinesia has further been proved by PET studies in humans showing an elevated D3 receptor binding in patients with dyskinesia [49][50]. It has been reported that D3 receptor agonists may produce neuroprotective effects by directly scavenging free radicals, improving the activity of free radical scavenging enzymes, stabilizing the mitochondrial membrane, directly inhibiting neuronal apoptosis. Moreover, being D3 receptors primarily localized in the midbrain limbic system, which is unrelated to motor function, selective D3 receptor agonists may have suitable anti-PD activity without significant extrapyramidal side effects [50][51][52][53][54][51,52,53,54,55].
The D3 receptor subtype has also been shown to exhibit biased signaling and desensitization pattern in response to certain agonists, DA included. Such an evidence could significantly contribute to the development of motor and hyperkinetic symptoms in PD and LID, respectively. On the contrary, the closely related D2 receptors have not demonstrated these D3 characteristics [39][55][40,56]. Thus, it has been demonstrated that the selective D3 agonist SK609, which does not induce desensitization of D3 receptors in vivo [56][57][57,58], was able to decrease locomotor activity [58][59][59,60]. Moreover, it has also been observed a dose dependent efficacy of SK609 in improving motor deficits in PD and ameliorating abnormal involuntary movements (AIMs) in LID using the hemiparkinsonian unilateral lesioned rodent PD model. A combination of SK609 and a low dose of LD induced a motor symptomatic relief without producing AIMs [60][61].
CJ-1639 is actually one of the most potent and selective D3 full agonist reported to date that may become one of the newer anti-PD drugs [61][62][62,63].
The novel ‘multifunctional’ D2/D3 high-affinity compound D-512, endowed with receptor agonist activity together with antioxidant and other neuroprotective features has recently been developed [63][64][64,65]. Compared with Ropinirole, it showed greater peak-dose efficacy and a longer lasting action, thus deserving consideration for clinical investigation.
The novel carbazole-based multifunctional D2/D3 receptor ligands D-636, D-653, and D-656, endowed with high binding affinity and full agonist activity at both receptors [65][66], have been proved to be highly efficacious in a PD rat model indicating their potential in relieving motor dysfunction in PD. They also exhibited neuroprotective property in an in vitro cellular model of PD. Furthermore, D-636 and D-653 demonstrated potent modulator effect on aggregation and toxicity of α-synuclein protein in vitro. Thus, it has been postulated that multifunctional drugs like D-636, D-653, and D-656 have the potential to alleviate motor dysfunction in PD patients, as well as to modify the disease progression.
Pardoprunox (SLV-308), a D2/D3 receptor partial agonist and 5-HT1A receptor full agonist, reached Phase III clinical trials for the treatment of PD. Compared with other dopaminergic agents, it displayed lower propensity to elicit side effects like dyskinesia [66][67].
Since ligands endowed with such a multitarget profile might be effective in PD pharmacotherapy, novel multitarget compounds based on the N-((6,6-diphenyl-1,4-dioxan-2-yl)methyl)-2-phenoxyethan-1-amine (DDMPA) scaffold were studied. Interestingly, the 3-hydroxy derivative, here named for the first time DDMPA-8, behaved as a partial agonist at D2 and as a potent full agonist at D3 and D4 subtypes. In addition to its potent 5-HT1A receptor agonism, that might be helpful in reducing dyskinetic side effects associated with the dopaminergic stimulation, such a dopaminergic profile makes DDMPA-8 a potential multitarget compound for the treatment of PD. In perspective, its evaluation in PD animal models would shed light on its therapeutic potential [67][68].
D4 receptors are present within the basal ganglia that represent a key area involved in parkinsonism and, in particular, in dyskinesia [68][69][69,70]. During the last years, a renewed interest has emerged around D4 receptors as potential therapeutic target for the treatment of PD, in which D4 antagonists can attenuate LID [70][71][72][73][71,72,73,74]. It has been observed that associating the potent D4 antagonist L-745,870 to LD significantly ameliorates the dyskinesia scenario in LID models. Such a result was quite remarkable since this compound has also demonstrated to be well tolerated in clinical trials. Thus, it could have a rapid development as a new tool for LID treatment. Unfortunately, disappointing results were obtained in the rotarod performance test when co-administered with LD. In fact, L-745,870 reduced the overall LD antiparkinsonian benefit in this model opening only a narrow therapeutic window to its use for the treatment of LIDs [74][75][75,76].
The effect of the novel selective D4 antagonist, VU6004461 [76][77], endowed with high blood–brain barrier penetrability has also been investigated. The clear antidyskinetic effect of both L-745,870 and VU6004461 points to the D4 as a possible future target for the treatment of LID [77][78]. At present more work is needed, but the use of D4 antagonists for the treatment of LIDs in PD remains a very promising area of research and the development of more highly optimized ligands is still an acceptable challenge [72][73].
All the results obtained so far are not enough and the rising of the aged population imposes new strategies in PD that may help to manage known limitations of current therapies. Some of the alternative strategies investigated as potential treatment of LID in PD involve non-dopaminergic receptors. To help researchers in such a challenge, this review focuses on recent investigations about non-dopaminergic CNS receptor ligands that have been identified to have therapeutic potential for the treatment of motor and non-motor symptoms of PD. Such agents in different way may contribute to extend LD response and/or ameliorate LD-induced side effects.
2. Serotonin Receptors
The serotonin (5-HT) system has been demonstrated to play a crucial role in the pathogenesis of LID in animal models of PD [78][79]. Indeed, after advanced dopaminergic cell loss, remaining serotonin neurons can convert exogenous LD to DA and mediate its vesicular storage and release [37][79][38,80]. The non-physiological DA release from these neurons might cause DA receptor overstimulation, leading to generation of dyskinesia [80][81]. Consequently, modulation of 5-HT system has emerged as a promising strategy for LID management. Several studies have shown a reduction of LID induced by targeting different 5-HT receptor (5-HTR) subtypes. 5-HT1AR (dorsal raphe nucleus and striatum), 5-HT1BR (striatopallidal pathways), and 5-HT2AR (substantia nigra pars reticulata and internal segment of the globus pallidus) can modulate DA, GABA, and glutamate release within the basal ganglia to improve motor symptoms of PD and to reduce dyskinesia [81][82]. 5-HT1AR and 5-HT1BR agonists, as well as 5-HT2AR and 5-HT3R antagonists have demonstrated a potential as antidyskinetic agents, while 5-HT4 agonists can increase LD-stimulated DA release in CNS.2.1. 5-HT1ARs
5-HT1AR is the most studied of the 5-HT family. Indeed, several preclinical and clinical studies demonstrated that 5-HT1AR stimulation (auto- and heteroreceptors) [48][82][83][49,83,84] may reduce dyskinesia through the decrease of DA release [78][79]. Moreover, 5-HT1AR activation may also weaken glutamatergic transmission ameliorating motor symptoms [82][83].
In a preclinical study, the highly selective 5-HT1A full agonist 8-OH-DPAT and its (R)-(+) eutomer reduced LID, but also worsened motor function in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-lesioned primate model [84][85] (Figure 3).
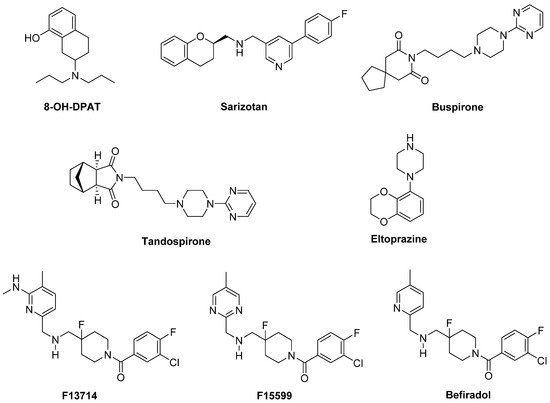
Figure 3. 5-HT1AR agonists.
Sarizotan, another 5-HT1AR full agonist, also endowed with partial D2-like agonist/antagonist profile [85][86], showed better results as an antidyskinetic agent in a 6-hydroxydopamine (6-OHDA)-lesioned rat model of PD [86][87]. When evaluated in a Phase II clinical study, at low dose it ameliorated some PD symptoms [87][88]. However, it failed in attenuating LID compared with placebo in two Phase III studies [88][89], limiting its therapeutic potential.
The partial 5-HT1AR agonists Buspirone and Tandospirone have been shown to possess antidyskinetic properties in humans, but also negatively impacted on parkinsonian symptoms [89][90][90,91]. Currently, a Phase III clinical trial is investigating the antidyskinetic potential of Buspirone, which also behaves as a D2-like receptor antagonist [91][92], on LID (NCT02617017), while a Phase I clinical trial is exploring its potential in combination with the non-selective NMDA antagonist Amantadine (NCT02589340).
Another 5-HT1AR partial agonist able to reduce LID in combination with LD in preclinical studies is Eltoprazine [92][93]. Unlike Buspirone and Tandospirone, this compound also behaves as a 5-HT1BR agonist and a 5-HT2CR antagonist [93][94]. In both rodent and monkey models, it abolished LD-mediated motor improvements, suggesting that it may have a narrow therapeutic window [92][93]. However, the loss of LD efficacy proved to be mitigated by co-administration with 5-hydroxy-tryptophan [94][95]. When tested in a clinical Phase I/IIa study, Eltoprazine attenuated LID without affecting the antiparkinsonian action of LD [95][96]. To further validate its efficacy, another Phase II trial is currently ongoing to assess the duration of Eltoprazine’s efficacy in LID management and its effects on motor function (NCT02439125). Preclinical studies have also highlighted the potential efficacy of combining eltoprazine with other compounds able to attenuate LID, such as Amantadine and the selective adenosine A2A receptor antagonist Preladenant [92][96][97][93,97,98].
The 5-HT1A agonists so far evaluated in clinical trials have shown off-target effects and only partial agonist efficacy at 5-HT1AR. In this contest, the new highly selective 5-HT1AR biased agonists F13714, F15599, and Befiradol (also known as F13640 or NLX112) were recently demonstrated to exhibit exceptionally potent antidyskinetic activity in animal models of PD, while minimally interfering with LD antiparkinsonian effects [98][99][100][99,100,101]. Biased 5-HT1A agonists are selective ligands that act in specific brain regions and preferentially target different 5-HT1AR subpopulations [101][102]. While F13714 and Befiradol preferentially bind presynaptic 5-HT1A autoreceptors, F15599 activates postsynaptic 5-HT1ARs [99][102][103][100,103,104]. Befiradol has recently been shown to possess a distinctive in vitro G-protein activation profile in rat brain cell membranes which differs from those of F13714 or F15599 [104][105]. In particular, it preferentially activate Gαo proteins over other G-protein subtypes. This compound is currently undergoing clinical development as an antidyskinetic agent (www.parkinsons.org.uk/news/investing-new-treatment-dyskinesia).
3. Glutamate Receptors
In rodent models of LID, high extracellular levels of glutamate were observed in the striatum and substantia nigra pars reticulata. Molecular imaging studies suggested that similar neurochemical changes of this system are evident in PD patients [105][134]. Therefore, glutamate receptors represent attractive targets for the treatment of LID. While the first efforts were addressed to antagonize the ionotropic glutamate receptors (iGluRs) N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) subtypes [106][135], more recently, metabotropic glutamate receptors (mGluRs) have also been considered as potential targets for PD and LID treatment [107][108][136,137].3.1. iGluRs
Several studies performed in animal models of LID and in post-mortem basal ganglia tissues from dyskinetic PD patients have revealed modifications in the expression and state of phosphorylation of iGluRs, in particular NMDA and AMPA receptors [105][134]. Therefore, these receptor systems are considered of major importance to the pathophysiology of LID.
3.1.1. NMDA Receptors
Alterations in NMDA receptor trafficking and distribution in the postsynaptic neurons appear to be associated with the extent of DA denervation as well as with the development of LID. However, the exact mechanisms regulating NMDA receptor subcellular trafficking and function in PD and LID are not fully elucidated yet [109][110][138,139]. Among the subunits forming the NMDA receptor, GluN2B subunit has attracted considerable interest. Indeed, from radioligand binding studies, performed both in NHP models of LID and dyskinetic PD patients, increased binding densities at GluN2B-containing NMDA receptors in the putamen were observed [111][112][140,141]. Furthermore, increased levels of GluN2B phosphorylation have been found in 6-OHDA-lesioned rats after chronic LD treatment [113][142].
The GluN2B-selective antagonists Ifenprodil and Traxoprodil (CP-101606) were reported to ameliorate parkinsonian symptoms and to reduce LID in rat and NHP models [114][115][116][117][118][119][143,144,145,146,147,148] (Figure 5). However, their use has been discouraged in NHP owing to the development of severe side effects, including amnesia and dissociation [114][116][120][121][143,145,149,150].
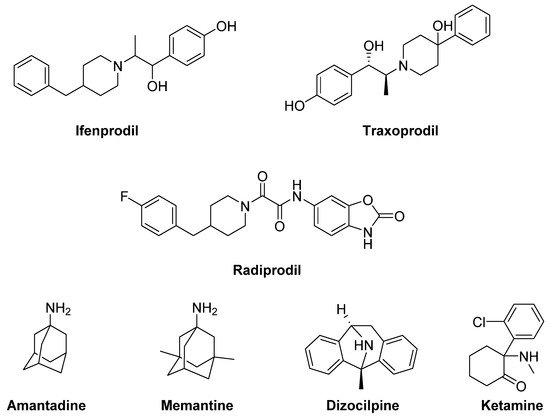
Figure 5. N-methyl-D-aspartate (NMDA) receptor ligands.
In general, divergent results have been obtained following treatment with GluN2B-selective antagonists in animal models of LID, ranging from improvement to no effect, and even to a worsening of AIMs [105][134].
Radiprodil, another GluN2B-selective antagonist, in combination with the selective A2A receptor antagonist Tozadenant, significantly improved motor activity both in 6-OHDA-lesioned rats and MPTP-lesioned NHP models, suggesting that the use of such a combination could lead to motor improvement to PD patients, without inducing the motor complications induced by LD therapy [122][123][151,152].
Promising results for the treatment of LID were also obtained with the weak non-competitive NMDA receptor antagonists Amantadine and Memantine. Amantadine, historically used as an antiviral agent, showed moderate but significant antidyskinetic efficacy in various clinical trials performed in the last two decades [124][125][126][153,154,155]. For this reason, it is the only drug with established antidyskinetic activity available in the market [127][156]. Amantadine treatment proved to reduce the duration of LID and to improve motor disability in PD [128][157] without major complications [125][154]. However, there are contrasting results concerning its long-term efficacy [129][130][158,159]. A Phase II clinical trial is currently ongoing to study the impact of Amantadine in preventing LID in early PD (NCT01538329). Other Phase II clinical trials are currently underway to evaluate the efficacy of Amantadine, in combination with other classes of drugs (e.g., Buspirone or Eltoprazine, see the section “Serotonin receptors”), in reducing LID in preclinical or clinical trials. Moreover, a recent study has revealed that the combination of a sub-effective dose of Amantadine and the nitric oxide synthase inhibitor 7-Nitroindazole potentiated the effect of reducing LD-induced AIMs in 6-OHDA-lesioned rats when compared to the effect of the drugs alone. This strategy may provide therapeutic benefits to PD patients at lower and thus more tolerable doses [131][160]. Memantine has also been investigated for its antidyskinetic potential in PD patients, but the results were conflicting. Although Memantine treatment was associated with lower LID scores and reduced daytime duration of dyskinesia, no significant effects on dyskinesia severity were found [132][133][134][161,162,163].
Other non-competitive NMDA receptor antagonists, including Neu-120 (structure not disclosed), Dizocilpine (MK-810) and Ketamine, displayed potential antidyskinetic effects.
Neu-120 is produced by Neurim Pharmaceuticals for the treatment of drug-induced dyskinesias. This compound, that also inhibits MAO-B and GSK-3β, has been subjected to a Phase I/II clinical trial to determine its safety, tolerability, pharmacokinetic and pharmacodynamic profiles in reducing LID in patients with advanced-phase idiopathic PD (NCT00607451). The study has been completed, but the results are not available yet.
Dizocilpine also reduced LD-induced AIMs in a rat model of LID, but only at concentrations that worsen parkinsonism [135][164]. However, when this compound was co-administered with the opioid glycopeptide Lactomorphin (see Figure 17), its pro-parkinsonian activity was suppressed, while a strong antidyskinetic effect remained [136][165].
Finally, the dissociative anesthetic Ketamine, administered at low sub-anesthetic doses, displayed a long-term effect in reducing LID in a preclinical 6-OHDA-lesioned rat model [137][166]. This result was confirmed by a clinical trial, in which intravenous infusion of low doses of Ketamine induced a long-lasting therapeutic benefit to reduce LID and depression in PD patients [138][167].
3.1.2. AMPA Receptors
Analogously to NMDA receptors, synaptic localization and phosphorylation of AMPA receptors proved to be altered in animal models of LID and in PD patients [139][140][141][142][168,169,170,171]. Moreover, in MPTP-lesioned monkeys and 6-OHDA-lesioned rats, the pharmacological blockade of AMPA receptors decreased LIDs and enhanced the antiparkinsonian effect of LD [143][144][145][172,173,174]. Conversely, AMPA receptor agonists triggered dyskinesias [145][174]. In the light of these findings, treatments with selective AMPA receptor antagonists alone or in combination with selective NMDA receptor antagonists showed beneficial effect in reducing dyskinesia [143][172].
The anticonvulsants Topiramate and Perampanel are the only AMPA receptor antagonists which have reached clinical trials (Figure 6). Topiramate is a negative modulator of AMPA receptors [146][175] and a PAM of GABAA receptors [147][176]. It has been reported to improve LID in MPTP-lesioned NHPs [148][177]. Moreover, in combination with the non-competitive NMDA receptor antagonist Amantadine, Topiramate elicited a synergistic antidyskinetic effect in both rodent and marmoset models of LID at low doses [149][178]. Despite these positive preclinical experiences, clinical trials have provided conflicting results. In a double-blind trial involving patients with idiopathic PD, Topiramate worsened dyskinesia and was poorly tolerated [150][179]. No results are so far available for other two Phase II clinical trials evaluating the efficacy of the combination of Amantadine and Topiramate versus Amantadine alone in PD patients with or without dyskinesia (NCT00794313, NCT01789047). Conflicting results were also found in clinical trials with the non-competitive antagonist Perampanel [151][152][153][180,181,182].
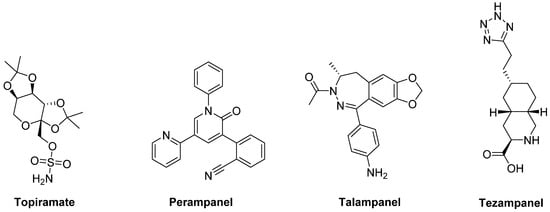
Figure 6. amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor ligands.
In preclinical studies, Talampanel (LY-300164, GYKI 537773), another non-competitive AMPA receptor antagonist, increased the anti-parkinsonian benefits of LD in MPTP-treated monkeys [145][174], while the competitive AMPA antagonist Tezampanel (LY-293558) was able to reduce wearing-off of LD-induced motor responses in 6-OHDA-lesioned rats [154][183].
4. Noradrenergic Receptors
The noradrenergic system plays an important role in the pathophysiology of PD. Noradrenergic neurons in the locus coeruleus [155][210] undergo degeneration in PD and may even anticipate the death of DA neurons [156][157][158][211,212,213]. They appear to play a protective role by establishing the extent of nigral degeneration induced by both neurotoxic damage and pathological events underlying PD [157][159][160][212,214,215]. Therefore, the indirect activation of adrenergic pathways by blocking presynaptic α2 adrenergic autoreceptors should prevent the nigrostriatal DA degeneration and subsequent motor deficits in PD. Moreover, being the noradrenergic system implicated in autonomic function, targeting α2 or β adrenergic receptors (α2-Ars or β-Ars, respectively) appears to have potential to improve symptomatic orthostatic hypertension in PD.4.1. α2-Ars
Stimulation of α2-Ars overexpressed in striatal GABAergic neurons activates direct basal ganglia pathway and is involved in the generation of LID, justifying the investigation of α2-AR antagonists as antidyskinetic agents [161][216].
The non-selective α2-AR antagonist Idazoxan was effective in alleviating the expression of AIMs in 6-OHDA-lesioned rats [162][217] (Figure 10). In a randomized, placebo-controlled pilot study, Idazoxan improved the severity of LIDs without affecting the antiparkinsonian effect of LD [163][218], but increasing the frequency of cardiovascular side effects.
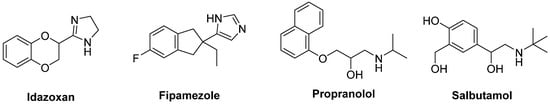
Figure 10. Noradrenergic receptor ligands.
Fipamezole, a more recently developed α2-AR antagonist, has also been shown to extend both the duration and quality of LD action in MPTP-lesioned NHP [164][219]. A clinical trial with ten PD patients has demonstrated good tolerability and sound antidyskinetic effect [165][220]. In a Phase II double-blind, placebo-controlled study in US and Indian PD patients Fipamezole did not show any significant antidyskinetic effect. However, the analysis of US subjects revealed that it reduced LIDs in a dose-dependent manner with an acceptable profile of adverse effects [166][221]. Other clinical trials with Fipamezole have been performed but the results have not been published yet (NCT01149811, NCT01140841, NCT00040209).
4.2. β-ARs
Pharmacological and neuroanatomical evidences support a role for β-ARs as potential therapeutic targets against LID. Indeed, both β1- and β2-ARs are expressed in the striatum [167][222] and are integral in PD patients [168][223].
The β2-AR antagonist Propranolol has been reported to reduce LID without affecting LD’s efficacy in several experimental and clinical studies (Figure 10). However, it failed to reduce dyskinesia produced by the D1 receptor agonist SKF81297 or the D2 receptor agonist Quinpirole. Antidyskinetic properties of Propranolol appear to be mediated via attenuation of LD-induced extra-physiological efflux of DA [169][224]. β blockers might be preferred first-line agents in PD patients who has co-morbid hypertension. Moreover, they are associated with a lower risk of constipation, which is one of the most frequent non-motor symptoms of PD [170][225]. However, in patients with asthma or chronic obstructive pulmonary disease, β blockers should not be used owing to the risk of bronchospasm [171][226].
Evidences also support the use of β2-AR agonists in PD therapy. Indeed, from molecular and immunological studies adrenergic stimulation has been suggested to decrease both α-synuclein deposition and release of neurotoxic molecules. In small clinical trials the β2-AR agonist Salbutamol in combination with LD improved parkinsonian symptoms in patients with fluctuating PD. Nevertheless, large randomized controlled trials are lacking [172][227].